Effects of Paraneoplastic LEMS on Neuromuscular Junction
Effects of Paraneoplastic Lambert-Eaton Myasthenic Syndrome on the Neuromuscular Junction
Â
Introduction
The Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune disease. It is characterised by muscle weakness of the proximal lowerlimbs as a result of auto-antibodies produced against the voltage-gated calcium channels (VGCC) found on the presynaptic membrane of the neuromuscular junction(1). Most of the time, LEMS is classified as a paraneoplastic syndrome as it is often associated with cancer (small cell lung cancer in particular) (2, 3).
One of the earliest descriptions of this disease was reported by Anderson and his colleagues in the year 1953, describing a 47-year-old patient with bronchial carcinoma presenting with progressive muscle weakness(4). Upon examining this patient, Anderson wrote:
His (the patient’s) muscle power was generally weaker than his muscle bulk would suggest, especially the muscles of the shoulder girdle, hip girdle, trunk, and anterior compartments of the legs.(4)
Since then, much research has been done to find out more about this disease. It is now known that the auto-antibodies are primarily directed against the P/Q-type VGCCs at the neuromuscular junction (1-3, 5, 6). This report will discuss the effects of LEMS on the neuromuscular junction.

 Voltage-Gated Calcium Channels
Voltage-Gated Calcium Channels
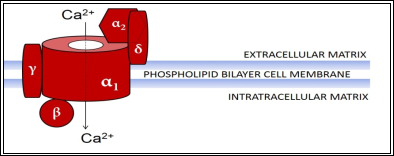
It would be useful to first understand the structure of the voltage-gated calcium channels (VGCC). In general, VGCCs are made up of 5 subunits (α1, α2, β, δ, γ) and can be divided, according to the electrophysiological and pharmacological properties of their α1 subunit, into 5 different subtypes (L, N, P/Q, R and T-type)(2, 7). The structure of a typical VGCC is shown in Figure 1 below.

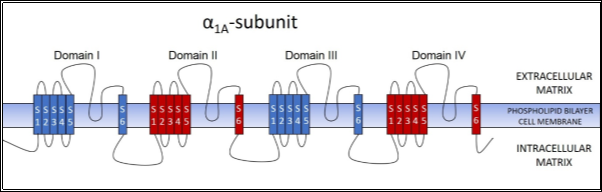 As mentioned before, the LEMS auto-antibodies are mainly directed against the P/Q-type VGCCs (1-3, 5, 6). The α1 subunit of the P/Q-type VGCCs is known as α1A(8). This α1A subunit forms the channel which allows the movement of Ca2+ ions into the synaptic knob(8), and can be further broken down into 4 identical domains (I, II, III, IV), each possessing 6 transmembrane segments (S1-S6)(2, 8) (see Figure 2).
As mentioned before, the LEMS auto-antibodies are mainly directed against the P/Q-type VGCCs (1-3, 5, 6). The α1 subunit of the P/Q-type VGCCs is known as α1A(8). This α1A subunit forms the channel which allows the movement of Ca2+ ions into the synaptic knob(8), and can be further broken down into 4 identical domains (I, II, III, IV), each possessing 6 transmembrane segments (S1-S6)(2, 8) (see Figure 2).
What Happens at The Neuromuscular Junction?
Now that we have understood the basic structure of the VGCCs, let us move on to its role and how it is affected by the pathogenesis of LEMS. The neuromuscular junction is made up of the pre-synaptic membrane, the synaptic cleft and the post-synaptic membrane (5, 9, 10). P/Q-type VGCCs present on the surface of the pre-synaptic membrane play a vital role in allowing synaptic transmissions to cross the neuromuscular junction (7, 9).
In studies conducted by multiple different researchers, anti-P/Q-type VGCC auto-antibodies were found in more than 85% of LEMS patients (3, 6, 11). Japanese researchers Masaharu Takamori and his colleagues even went a step further to determine the specific immunodominant sites within the P/Q-type VGCC in which the auto-antibodies would bind. In their study, they found that majority of their test subjects had auto-antibodies directed against domains II and IV of the α1A subunit in the P/Q-type VGCC(12)(highlighted in red in Figure 2).
Depolarisation of the pre-synaptic membrane occurs when stimulated by an action potential initiated by a nerve impulse(13). This membrane depolarisation causes the VGCCs to open, allowing the local influx of calcium ions (Ca2+)into the synaptic knob(13). The increase in Ca2+ concentration will stimulate the exocytosis of vesicles containing the neurotransmitter Acetylcholine (ACh) into the synaptic cleft(13). ACh then goes on to bind to the nicotinic acetylcholine receptors found on the motor end plate which will trigger an action potential and subsequently causing muscle contraction.(10, 13)
In patients with LEMS, the LEMS auto-antibodies would bind to the VGCC present on the pre-synaptic membrane of the neuromuscular junction(5). This causes the VGCC to lose its ability to function as an ion channel, thus inhibiting the influx of Ca2+ into the synaptic knob during membrane depolarisation(5). Since the exocytosis of synaptic vesicles are dependent on Ca2+ (14), it is believed that a decrease in Ca2+ influx would result in a reduction in synaptic vesicle exocytosis(5, 13, 15).
A recent study conducted in 2015 confirmed the phenomenon in which the LEMS IgG directly causes a reduction in the exocytosis of synaptic vesicles (16). They did this by using fluorescence imaging techniques to observe the exocytosis of synaptic vesicles in rat neurons incubated with LEMS IgG (16). Pooled IgG from healthy individuals were used in comparison and as a control (16).
Hence, it is evident that the muscle weakness seen in LEMS patients is a direct consequence of reduced exocytosis of ACh-containing synaptic vesicles.
In summary, the Lambert-Eaton myasthenic syndrome is an autoimmune disease that causes muscle weakness in patients. The reduction in muscle power is due to insufficient ACh release at the neuromuscular junction. In 85% of these patients, this is caused by auto-antibodies inactivating the P/Q-type VGCCs on the presynaptic membrane.
Going Beyond VGCCs
So, what happens in the remaining 10-15% of LEMS patients that do not have anti-P/Q-type VGCC antibodies? Many scientists have asked the same question and investigations have been carried out to find alternative mechanisms in the pathogenesis of LEMS in seronegative patients. (The term “seronegative” is used to describe patients with undetectable anti-P/Q-type antibodies.)
In the absence of anti-P/Q-type VGCC antibodies, it is almost impossible that LEMS would have the same pathogenesis in seronegative patients. Yet, it is found that there are no significant differences in the electrophysiological and clinical characteristics between the seronegative and seropositive patients(17, 18).
One possible explanation for this phenomenon is that there might be auto-antibodies directed against a different molecule involved in the synaptic transmission; hence causing seronegative patients to have the same clinical and electrophysical features albeit having no anti-P/Q-type VGCC antibodies.
To date, many auto-antibodies with different target molecules have been found in LEMS patients. One of the earliest to be discovered is an auto-antibody to synaptotagmin, which is a protein involved in synaptic vesicle exocytosis (2, 5, 13, 15, 19).
Furthermore, antibodies to the M1-type presynaptic muscarinic ACh receptor (M1-mAChR) have also been discovered(20). The M1-mAChR is a G-protein coupled receptor that regulates ACh release at the neuromuscular junction(5, 20). We will not be going into the specifics of these antibodies in this report as the mechanisms involved are largely complicated.
Conclusion
Even though LEMS is a rare autoimmune disease, the prevalence of LEMS as a paraneoplastic syndrome in patients with small cell lung cancer (SCLC) is extremely high. More than half of LEMS patients have SCLC and more often than not, the diagnosis of LEMS precedes the diagnosis of SCLC(21). Therefore, LEMS could potentially play a crucial role in allowing certain cancers to be detected at an earlier stage.
Hence, understanding the pathophysiology of this disease would not only prove useful in discovering better treatments for LEMS patients but also aid the early detection of cancer.
References:
1.Weiss N, Koschak A. Pathologies of Calcium Channels. Weiss N, Koschak A, editors: Berlin, Heidelberg : Springer Berlin Heidelberg : Imprint: Springer; 2014.
2.Takamori M. An autoimmune channelopathy associated with cancer: Lambert-Eaton myasthenic syndrome. Intern Med. 1999;38(2):86-96.
3.Lennon VA, Kryzer TJ, Griesmann GE, O’Suilleabhain PE, Windebank AJ, Woppmann A, et al. Calcium-Channel Antibodies in the Lambert-Eaton Syndrome and Other Paraneoplastic Syndromes. New England Journal of Medicine. 1995;332(22):1467-75.
4.Anderson HJ, Churchill-Davidson HC, Richardson AT. BRONCHIAL NEOPLASM WITH MYASTHENIA. The Lancet. 1953;262(6799):1291-3.
5.Hulsbrink R, Hashemolhosseini S. Lambert-Eaton myasthenic syndrome – diagnosis, pathogenesis and therapy. Clin Neurophysiol. 2014;125(12):2328-36.
6.Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Journal of the Neurological Sciences. 1997;147(1):35-42.
7.Catterall WA. Voltage-gated calcium channels. Cold Spring Harbor perspectives in biology. 2011;3(8):a003947.
8.Structure and Regulation of Voltage-Gated Ca2+ Channels. Annual Review of Cell and Developmental Biology. 2000;16(1):521-55.
9.Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle & Nerve. 2006;33(4):445-61.
10.Martini F, Nath JL, Bartholomew EF. Fundamentals of anatomy & physiology. San Francisco: Benjamin Cummings; 2012.
11.Motomura M, Johnston I, Lang B, Vincent A, Newsom-Davis J. An improved diagnostic assay for Lambert-Eaton myasthenic syndrome. Journal of Neurology, Neurosurgery & Psychiatry. 1995;58(1):85-7.
12.Takamori M, Iwasa K, Komai K. Antigenic Sites of the Voltage-gated Calcium Channel in Lambert-Eaton Myasthenic Syndromea. Annals of the New York Academy of Sciences. 1998;841(1):625-35.
13.Lang B, Newsom-Davis J. Immunopathology of the Lambert-Eaton myasthenic syndrome. Springer seminars in immunopathology. 1995;17(1):3-15.
14.Mechanisms of Synaptic Vesicle Exocytosis. Annual Review of Cell and Developmental Biology. 2000;16(1):19-49.
15.Takamori M. Lambert-Eaton myasthenic syndrome as an autoimmune calcium channelopathy. Biochem Biophys Res Commun. 2004;322(4):1347-51.
16.Spillane J, Ermolyuk Y, Cano-Jaimez M, Lang B, Vincent A, Volynski KE, et al. Lambert-Eaton syndrome IgG inhibits transmitter release via P/Q Ca2+ channels. Neurology. 2015;84(6):575-9.
17.Oh SJ, Hatanaka Y, Claussen GC, Sher E. Electrophysiological differences in seropositive and seronegative Lambert-Eaton myasthenic syndrome. Muscle & Nerve. 2007;35(2):178-83.
18.Nakao YK, Motomura M, Fukudome T, Fukuda T, Shiraishi H, Yoshimura T, et al. Seronegative Lambert-Eaton myasthenic syndrome: study of 110 Japanese patients. Neurology. 2002;59(11):1773.
19.Takamori M, Hamada T, Komai K, Takahashi M, Yoshida A. Synaptotagmin can cause an immune-mediated model of Lambert-Eaton myasthenic syndrome in rats. Annals of neurology. 1994;35(1):74-80.
20.Takamori M. Lambert-Eaton myasthenic syndrome: Search for alternative autoimmune targets and possible compensatory mechanisms based on presynaptic calcium homeostasis. Journal of Neuroimmunology. 2008;201-202:145-52.
21.Titulaer MJ, Verschuuren JJGM. Lambert-Eaton Myasthenic Syndrome. Annals of the New York Academy of Sciences. 2008;1132(1):129-34.