Human African Trypanosomiasis Treatment
|
THE synthesis of novel hydrazone compounds to determine the potential anti-parasitic properties and efficacy in the treatment of Human African Trypanosomiasis. ABSTRACT |
|
Human African Trypanosomiasis is caused by two sub-species of trypanosome, Trypanosoma brucei gambiense and Trypanosoma brucei rhodeisense. The protozoan parasite has complex mechanisms to evade immune destruction and hence survives in the host, leading to eventual death without treatment. Treatment such as Suramin and Melarsoprol are limited and have high toxicity, often leading to further complications. Alternative therapies are drastically needed to combat trypanosomal infection, with novel hydrazone compounds currently in development promoting trypanocidal activity. The aims and objectives of this research project include the synthesis of novel hydrazone compounds, with aims of high efficacy and low toxicity, namely against Trypanosoma brucei rhodesiense. This was achieved through a series of reactions, with proton NMR spectra used to confirm the structures of novel hydrazone compounds, and biological screening analysis used to assess the trypanocidal activity in vitro. IC50 values were obtained for all hydrazones synthesized, the best results coming from HD1, 9.29 µM, and HD6, 16.18 µM. Overall, the presence of chlorine in the compound typically lowered trypanocidal activity when compared to HD1, which lacked chlorine atoms. Despite other compounds in literature having much lower IC50 values, the results remain promising, demonstrating trypanocidal activity against T. brucei brucei, the infective form of trypanosome that affects animals. Trypanocidal activity against Trypanosoma brucei brucei suggests potential activity against Trypanosoma brucei rhodesiense, signifying the IC50 values obtained from novel hydrazones. Furthermore, the biological properties of hydrazone structures demonstrate the potential to develop additional analogues and derivatives, which could contribute to combatting Trypanosoma brucei rhodesiense infection. Likewise, alternative therapies that can cross the blood-brain-barrier are another interest regarding research in the field. Trypanosoma brucei rhodesiense proceeds rapidly once an individual has been exposed to the parasite, with severe side effects and high mortality rates. This reinforces the need for better and more effective treatment and management of the disease. Key words: Trypanosomiasis, Trypanosoma brucei rhodesiense, hydrazone, Suramin, Melarsoprol, proton NMR, IC50 values |
1.1. Background of Human African Trypanosomiasis
Human African Trypanosomiasis (HAT), or African sleeping sickness, is a disease affecting a large proportion of Africa, with a suspected 60 million people at risk (Kennedy, 2013).It is caused by two subspecies of trypanosome, a unicellular and flagellated protozoan parasite, with Trypanosoma brucei rhodesiense (T.b.r) causing East- African sleeping sickness, and Trypanosoma brucei gambiense (T.b.g) causing West-African sleeping sickness. West African sleeping sickness is much more prevalent in terms of the whole continent, with 6228 cases reported in 2013. Incidence has decreased massively since 2000, with 25,865 reported cases, although this figure is suspected to be higher due to unreported disease incidence. It is the causative sub-species in 98% of cases. The other 2% of cases are classified as East-African sleeping sickness, with 86 cases reported in 2013, approximately 88% lower than in 2000 (709 cases reported) (Franco et al., 2014). Despite the low incidence of T.b.r infection, symptoms are severe and death occurs in almost all patients without treatment. Treatment for suchinfections are limited and have high toxicity, signifying the need for development.
Transmission of the protozoan parasite is via the vector, the tsetse fly. Infections with T.b.r proceed rapidly, with entry of infectious metacyclic trypomastigotes into the human bloodstream, lymphatic system and cerebrospinal fluid (CSF) following the initial bite, where proliferation occurs. The tsetse fly ingests a blood meal from an infected animal, with cattle and ungulates acting as reservoirs (Palmer & Wells, 2012), and hence ingests trypomastigotes. Various cell divisions and binary fission occur in the mid-gut of the fly to form procyclic trypomastigotes, and conformational changes occur to allow the trypomastigotes to bind to the salivary epithelia. Once attached, the parasite can once again replicate via asymmetric division (Pepin, 2014) to form metacyclic trypanosomes. It is in this way that a person becomes infected with T.b.r (Langousis & Hill, 2014).
1.2. Immunology
The causative parasite manifests in hosts due to the evasion of the immune system through antigenic variation, delaying the immune response and therefore allowing the parasite to complete its complex lifecycle (Stijlemans et al., 2016). The parasite expresses variant surface glycoproteins (VSGs) on its cell membrane via a glycophosphatidylinositol anchor to serve as a protective barrier. While the immune response induces antibody development against the VSG being expressed, the parasite can ‘switch’ VSG due to the large amount of VSG genes the genome possesses. This causes new antibodies to form against the newly expressed VSG, and the parasite continues to change VSGs to avoid destruction. Furthermore, the parasites alter their energy metabolism and internal structure, presenting further issues for the immune system (Stijlemans et al., 2016). T.b.r. is resistant to human trypanosome lytic factors (TLFs) containing apolipoprotein L1 (ApoL1), and this is due to the existence of the serum resistance protein (SRP) coded for by the SRA gene. This binds to TLF-1 and therefore prevents ApoL1-mediated lysis of the infected parasitic cells (Kennedy, 2013; Bart et al., 2015).
Kato et al. noted the upregulation of certain cytokines following T.b.r infection, namely IFN-γ, IL-10, IL-6 and TGF-β (Kato et al. 2015). Furthermore, these cytokines may play a key role in the inflammatory immune response. IL-6 and IL-10 were upregulated upon CSF examination of late stage patients, and those with CSF trypanosomes had higher levels of WBCs, positively correlating with IL-6 CSF levels. Despite this, no significant changes in levels of cytokines at different stages in HAT were noted.
Both subspecies of trypanosome cause non-specific inflammatory responses, resulting in non-specific symptoms. This demonstrates the difficulty in diagnosis and staging of T.b.r infection, and therefore poses a hindrance in regards to timely treatment (Lamour et al. 2015).
1.3. Symptoms
Symptoms, typically manifesting 1-3 weeks after bite, include myalgia, hyperplasia of the lymph nodes and spleen, and weight loss in the haemolymphatic stage, and there is clear central nervous system (CNS) involvement in later stages (Pepin, 2014; Kennedy, 2013). It is in this meningoencephalitic stage where patients often exhibit behavioural and motor disturbances including tremors, speech complications, anxiety, confusion, personality changes and others. Further complications may manifest, including ocular difficulties, acute renal failure, multi-organ failure and chronic lymphocytic meningoencephalitis. Death is highly likely to occur in those who do not receive treatment. Trypanosoma brucei produces an aromatic compound called tryptophol, and this induces sleep in humans. Other complications with sleep include irregular patterns of sleep or interrupted sleep, insomnia during the night and sleepiness during the daytime.
1.4. Current problem
Current pharmaceutical treatment including intravenous Suramin or Melarsoprol is ineffective and potentially toxic, with patients suffering with neurological dysfunction and post- treatment reactive encephalopathy. Co-administration with eflornithine and nifurtimox has been used to treat T.b.r infection also, but remains somewhat ineffective.
Suramin is provided intravenously as the first line treatment for the haemolymphatic stage of T.b.r infection, however this treatment can lead to side effects such as renal failure, peripheral neuropathy and anaphylactic shock, amongst others. It acts by binding to enzymes in the glycosome and disrupts glycolysis within the trypanosome (Babokhov et al., 2013). Should the parasite cross the blood-brain barrier in the later stages of the disease, the treatment options are toxic and limited to the arsenic compound, Melarsoprol, which acts by disrupting trypanosomal redox mechanisms. Treatment with this can lead to further complications such as post-treatment reactive encephalopathy seen in 10% of patients (Palmer & Wells, 2012), subsequently causing comas, seizures and cerebral oedema for example.
Co-administration of eflornithine with nifurtimox, a hydrazone of 5-nitro-2-furaldehyde, has been shown to demonstrate trypanocidal activity, and is routinely used to treat T.b.g. It has shown trypanocidal activity against T.b.b in mice, however has been relatively ineffective against T.b.r. Furthermore, although eflourthrine abides Lipinski’s rule of five in theory, in practice it is a highly hydrophobic compound, and is therefore unlikely to cross inner membranes once administered (Gilbert, 2014). It is therefore administered intravenously.
Due to there being no self-cure for T.b.r, treatment is required for a recovery to be made. Furthermore, many of the drugs developed are only applicable to bloodstream trypomastigotes, rather than those that have crossed the blood-brain barrier (Palmer & Wells, 2012). Factors such as this demonstrate the problematic nature of the disease, as well as the need for alternative therapies to combat the infection.

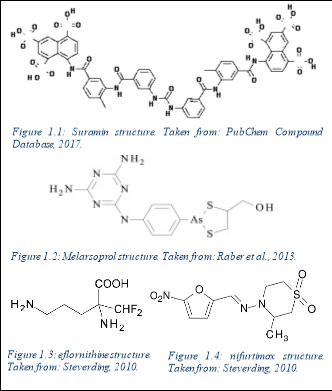
1.5. Current research
Currently, novel treatments are in development with the hope that toxicity is reduced and efficacy is increased against trypanosomal parasites, namely T.b.r and T.b.b.
To determine whether a proposed therapeutic compound is likely to be membrane permeable and therefore orally bioavailable, the Lipinski’s rule of 5 is applied. To be within the limits of the rule means that the compound is orally bioavailable and easily absorbed by the body (Leeson, P. 2012). The rules state that the molecular weight of the compound must be less than 500 Daltons, the lipophilicity value is less than 5, represented as a LogP value, the number of hydrogen bond acceptors must be less than 10, and the number of hydrogen donators must be less than 5. Hydrogen bond acceptors include highly electronegative atoms not bound to a hydrogen atom and with free electrons on its outer shell, including oxygen and nitrogen. In newer literature, fluorine may be considered and counted as a hydrogen acceptor. This contrasts with a hydrogen bond donor, any atom that donates a hydrogen atom that it is bound to break or form a bond. All novel compounds are assessed under these rules to determine properties involved in pharmacokinetics (absorption, distribution, metabolism and excretion). Oral administration is non-invasive, more bioavailable and simpler for patient use, all of which is desirable for a new treatment for T.b.r infection. Furthermore, a compound designed may still be orally bioavailable if a single rule is not adhered to.
Benzoxaborole compounds have recently been suggested as a new, novel treatment for Trypanosoma brucei (T.b.r and T.b.g) in vitro, namely SCYX-7158 oxaborole(Jacobs et al., 2011). The orally-active treatment is suggested to have high efficacy for both acute and chronic stages of the disease due to the treatment being permeable to the CNS, and properties such as distribution, metabolism, elimination, absorption and toxicology are apparent in vitro. A viability assay combined different whole cell T. brucei spp.and the novel compound to gain positive results of anti-parasitic activity. This treatment demonstrated concentration-dependency, and most trypanocidal activity came from the first 8 hours of a 24-hour exposure. Furthermore, the irreversibility of trypanocidal activity was noted during a short exposure. In vivo, SCYX-7158 was examined against an infection with T.b.b to monitor passage across the blood-brain barrier, with mice models providing an 80% cure rate over a 7 day-treatment. Benzoxaboroles, like hydrazones, have demonstrated anti-cancer, anti-fungal, anti- bacterial, anti-viral, anti-inflammatory and anti-parasitic properties with a low intrinsic toxicity similar to table salt when decomposed (Liu et al., 2014). This is due to the metabolites of benzoxaboroles being found to be boric acid and oxidative deboronation products, both with very low toxicity. Per Lipinski’s law, it is an orally bioavailable drug with very low IC50 values ranging from 0.19 to 1.008 µM, demonstrating high trypanocidal activity in vitro. An IC50 value is value is the inhibitory concentration value at which 50% inhibition of target cells has occurred. It is important when comparing the difference in the potency of compounds. A higher value represents less efficacy, with smaller values representing good trypanocidal activity in this case. It is a pharmacokinetic parameter, measuring the relationship between drug target and drug.
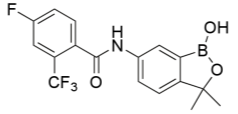

Other research at the forefront include the development of hydrazone compounds of 5-nitro-2-furaldehyde and adamantine alkanohydrazine (Foscolos et al., 2016). Adamantine and derivatives have been previously shown to have trypanocidal activity, increasing for the more hydrophobic, phenyl and cyclohexyl substituents. These compounds and derivatives demonstrated good trypanocidal activity, approximately 20 times greater than nifurtimox. The lowest IC50 value obtained was 0.386 µM. Following the same study, it was found that in the absence of the nitro group, no trypanocidal activity was demonstrated, highlighting the trypanosomal nitro reductase mechanism these novel compounds work by. In terms of the structure-activity relationship, it was seen that the selectivity of compounds against T. brucei species increased when the distance between the carbonyl group and the adamantine skeleton was increased, with derivatives containing 3-cyclopentyl and 3-phenyl being more biologically active than other compounds. In conjunction, the lipophilicity and conformational structure contributed to the efficacy of these novel compounds, with increased lipophilicity and conformational flexibility promoting trypanocidal activity.
1.6. Aims of research
Better pharmaceutical treatments with high efficacy and less lethal side-effects are drastically needed for T.b.r infection. Current research demonstrates the potential use of hydrazone compounds and derivatives in combatting parasitic infections, amongst others. Guidance from project supervisor Dr A Bhambra was given throughout the project regarding the structures of compounds. Hydrazones are defined as having the structure R1R2C = NNH2, the N=H bond is conjugated with a lone pair of electrons on the functional nitrogen atom (Verma et al. 2014). Research suggests hydrazone compounds also demonstrate anticancer, anti-inflammatory and anti-HIV properties, signifying biological variety and the potential to treat other diseases (Verma et al., 2014). It is because of these properties that new hydrazone compounds are in development, with the aim of synthesising novel compounds which demonstrate similar properties when applied against Trypanosoma brucei infection, and hence a potential therapeutic for T.b.r.
2.1. Chemistry/ experimental synthesis
The synthesis of four novel hydrazone compounds of reactants A-D and Pentafluorophenyl hydrazine (PFH) (Figure 3) was performed. All solvents and reactants were commercially available. Reflux condensation reactions with relevant reactant (3 mmol) and PFH (3 mmol) were performed, with continuous heating (oil bath) and stirring of compounds at 100-150 °C approximately. Compounds were separated with ethyl acetate (60 ml) and distilled water (50 ml). Excess magnesium sulphate was added to remove any excess water and products were vacuum filtered. Recrystallization reactions with boiling ethanol (10 ml approximately) not in excess were performed, and compounds were vacuumed down to give final compounds. Thin Layer Chromatography (TLC) analysis was performed for all compounds throughout the stages of experiment to monitor the constituents of the final product. Prior to NMR analysis, compounds were dissolved in chloroform (800 µL) with exception of HD6 (with reactant D), dissolved in DMSO (400 µL).
2.2. Biological Screening Analysis

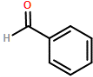
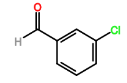 Further viability tests were carried out to assess the trypanocidal activity of each compound. Compounds were sent to the London School of Tropical Medicine for biological screening analysis against T.b.b. Stock drug solutions were prepared in DMSO at 20 mg/ml and further diluted appropriately. All assays were performed with use of 96-well microtiter plates, each well containing 100 µL of parasite culture, and serial dilutions of the compound in triplicate. Wells were incubated at 37°C for 72 hrs in 5% CO2. Control wells contained no compound, and blanks consisted of medium only. Following this, the MIC was determined and assurance of growth in control wells. 20 µL of Alamar Blue was added to wells, and plates were incubated for 2-4 hrs more. Plates were read on Gemini Plate Reader with an excitation wavelength of 530 nm and emission wavelength of 580 nm. Following this, IC50 values were obtained.
Further viability tests were carried out to assess the trypanocidal activity of each compound. Compounds were sent to the London School of Tropical Medicine for biological screening analysis against T.b.b. Stock drug solutions were prepared in DMSO at 20 mg/ml and further diluted appropriately. All assays were performed with use of 96-well microtiter plates, each well containing 100 µL of parasite culture, and serial dilutions of the compound in triplicate. Wells were incubated at 37°C for 72 hrs in 5% CO2. Control wells contained no compound, and blanks consisted of medium only. Following this, the MIC was determined and assurance of growth in control wells. 20 µL of Alamar Blue was added to wells, and plates were incubated for 2-4 hrs more. Plates were read on Gemini Plate Reader with an excitation wavelength of 530 nm and emission wavelength of 580 nm. Following this, IC50 values were obtained.

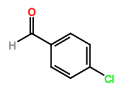


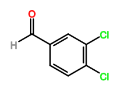

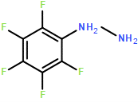

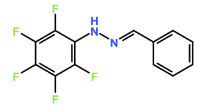
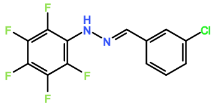
Four novel hydrazone compounds were synthesised, shown in table 1. The electrophilic carbonyl group of appropriate reactants A-D reacted with the nucleophilic nitrogen (hydrazine portion) of PFH by nucleophilic addition. IC50 values gained demonstrated potential, especially HD1 and HD6. Proton NMR analytical results from spectra obtained are demonstrated below, showing the amount of hydrogens (or protons) in each compound as well as their corresponding intensities. Throughout synthesis, TLC was performed for each compound to visualize the two reactants, the formed product and any other substance that may be present. Rf values and appropriate TLC analysis results are illustrated in alongside compounds in table 1.
3.1. HD1
Beige crystals (50% yield); δH (CDCl3) 7.28 (1H, s), 7.32-7.42 (3H, m), 7.64 (2H, dd), 7.82 (1H, s).
3.2. HD4
Pink crystals (50% yield) δH (CDCl3) 7.30 (2H, s), 7.35 (1H, s), 7.45-7.50 (1H, m), 7.62 (1H, s), 7.73 (1H, s).
3.3. HD5
Beige crystals (<25% yield); δH (CDCl3) 7.32-7.38 (3H, s), 7.54-7.60 (2H, d), 7.78 (1H, m).
3.4. HD6
Red crystals (<25% yield); δH (CDCl3) 7.37 (1H, s), 7.45 (2H, s), 7.70 (2H, s).
3.5. Biological screening analysis
Results obtained from the London School of Tropical Medicine included IC50 values for all hydrazones synthesised. These are listed alongside appropriate compounds in table 1. HD1 had an IC50 value of 9.29 µM; HD4 34.37 µM; HD5 34.09 µM and HD6 16.18 µM.
Formation of all compounds HD1, HD4, HD5 and HD6 (table 1) followed the general mechanism illustrated in appendix 1. R1 should be substituted with relevant reactant (A-D) benzene rings. Electrophilic reactants accept free pairs of electrons from the nucleophilic nitrogen atom of PFH by nucleophilic addition. The formation of a biologically active centre was documented (Verma, 2014). All compounds synthesised contain phenyl groups, which has been suggested in literature to promote trypanocidal activity (Foscolos et al., 2016). Hydrazones synthesised are also non-polar and hydrophobic compounds, meaning that they carry no net charge and are not soluble in water. Furthermore, an increase in hydrophobicity also promotes trypanocidal activity, however has been suggested to make the compound less bioavailable (Gilbert et al., 2014).
Proton NMR spectra demonstrate relative intensities in parts per million (ppm) of protons or hydrogens in the compound. Despite only hydrogens being of interest, it is important to consider the other electronegative groups that exist within the compound such as phenyl groups or -OH groups, which produce different peaks. The solvent peak demonstrates as a sharp, well distinguished peak to exclude it from the compound results. Singlet, doublet and
|
Name |
Reactant |
Structure |
IC50 value (µM) |
TLC (illustrated- not to scale) |
Rf value (cm) |
|
HD1 |
Benzaldehyde (reactant A) |
|
9.29 |
|
0.73 |
|
HD4 |
3-chlorobenzaldehyde (reactant B) |
|
34.37 |
|
0.48 |
|
HD5 |
4-chlorobenzaldehyde (reactant C) |
|
34.09 |
|
0.43 |
|
HD6 |
3,4-dichlorobenzaldehyde (reactant D) |
|
16.42 |
|
0.57 |

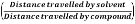







 multiplet peaks were all demonstrated on the spectra. NMR analysis reflects the structures synthesised, confirming the number and positioning of hydrogens. The spectra observed demonstrated some sharp peaks and others more broad. Furthermore, some peaks were somewhat distorted due to the presence of chlorine on the aromatic benzene ring.
multiplet peaks were all demonstrated on the spectra. NMR analysis reflects the structures synthesised, confirming the number and positioning of hydrogens. The spectra observed demonstrated some sharp peaks and others more broad. Furthermore, some peaks were somewhat distorted due to the presence of chlorine on the aromatic benzene ring.
Limitations of the experimental synthesis included low yields obtained from synthesised hydrazones. This could have been possibly due to the recrystallization step or some of the reactants not converting into product. Factors such as this could have been improved to gain better results. In addition, yields were estimated due to not weighing compounds beforehand, therefore weighing the compounds when they were initially synthesised would improve the data.
4.1. HD1
From the NMR spectra, HD1 contained 7 hydrogen atoms, one at 7.28 ppm as a singlet peak, three at 7.32 – 7.42 ppm as a mutliplet peak, two at 7.64 ppm as a doublet peak and one hydrogen at 7.82 ppm as a singlet peak. All novel compounds synthesised differ in terms of the aromatic benzene ring on the relevant reactants, therefore the positioning and amount of chlorine attached to this aromatic ring demonstrates the difference in IC50 values. This coincides with the biological activity against T.b.b. HD1 is formed of benzaldehyde and PFH, and therefore does not possess any chlorine atoms. The IC50 values were the lowest for HD1 at 9.29µM, which suggests that the presence of chlorine in the compound may decrease trypanocidal activity.
4.2. HD4
HD4 contained six hydrogen atoms, two at 7.30 ppm as a singlet peak, one hydrogen at 7.35 ppm as a singlet peak, one hydrogen at 7.45-7.50 ppm as a multiplet peak and a one hydrogen as a singlet peak at 7.73 ppm. HD4 contained a chlorine group on the 3rd carbon of the reactant benzene ring, with displacement of chlorine by (bio)nucleophiles, facilitated by the electrophilic carbon centre determining biological properties observed. However, it has been suggested that the presence of a chlorine atom at a non-reactive aromatic double bond diminishes reactivity (Naumann, 2003). The IC50 value for HD4 was 34.37 µM, the highest value from all the compounds synthesised. This supports the notion that chlorine atoms present in the compound reduces trypanocidal activity rather than improving it.
4.3. HD5
HD5 also contained six hydrogens, with three hydrogens at 7.32 – 7.38 ppm in a singlet peak, 2 hydrogens at 7.54 – 7.60 ppm in a doublet peak, and one hydrogen at 7.78 ppm in a multiplet peak. HD5 also contained a chlorine atom but on the forth carbon of the benzene ring, as opposed to the third. Altering the position of the chlorine atom did allow for an improved IC50 value to be obtained, which was 34.09 µM. This however remains much less effective than HD1.
4.4. HD6
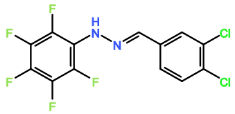
HD6 contained five hydrogen atoms, one at 7.37 ppm as a singlet peak, one at 7.35 ppm as a singlet peak, two at 7.45 ppm in a singlet peak, and 2 hydrogens at 7.70 ppm in a singlet peak. Despite the presence of chlorine in both HD4 and HD5 causing less effective activity against T.b.b, HD6 contained two chlorine atoms attached to the benzene ring, and the IC50 value obtained was nearly half that of HD4 and HD5 at 16.18 µM. This suggests that the presence of one chlorine atom existing at any carbon on the benzene ring does not promote trypanocidal activity, however the presence of 2 attached chlorine atoms did aid activity.
4.5. Oral bioavailability
Per Lipinski’s rule of 5, all novel compounds fall within the laws except from HD1, which contains 6 hydrogen bond donors. HD1 therefore would be more bioavailable if administered intravenously, however may still be orally bioavailable as only one rule has been violated, with the rest all being adhered to. Despite this, the other novel compounds alone demonstrate progression in seeking an orally bioavailable treatment for HAT, linked with reducing toxicity amongst being easier and more accessible for those who require it. Furthermore, the use of PFH incorporates many fluorine atoms, which increases lipophilicity (Citation), increasing activity in vivo, as well as increasing fat solubility and therefore allowing the compounds to easily pass through membranes in the body. Retaining fluorine is a desirable property due to the improvement of metabolic stability as a result of the high strength C-F bond. It is a relatively small molecule, with a high electronegativity value. All properties such as this contribute to biological responses within the body.
4.6. Comparison to literature IC50 values
HD1 and HD6 demonstrated the most promising results of 9.29 and 16.18 µM. For comparison intentions, alternative novel therapies at the forefront of science such as adamantine alkanohydrazine hydrazones and benzoxaborole SCYX-7158 have IC50 values of 0.386µM and 0.19 to 1.008µM respectively (Foscolos et al., 2016; Jacobs et al., 2011). Although the results from the synthesised compounds are promising and suggest trypanocidal activity against Trypanosoma brucei, both SCYX-7158 and the adamantine alkanohydrazine hydrazones have much lower IC50 values, suggesting that the structures of novel compounds could be improved and altered to gain better trypanocidal activity. Furthermore, current treatment Melarsoprol and Suramin have IC50 values of 0.046 and 0.004-0.009 µM, much lower than the compounds synthesised (Torreele et al., 2010). However, as stated above, these treatments offer high toxicity and high mortality rates following administration, and the fact that the novel compounds synthesised (HD1 especially) have low IC50 values remains promising. Furthermore, both Suramin and Melarsoprol are administered intravenously, posing more problems in terms of accessibility as developing countries where the disease is endemic possibly do not possess the facilities to administer treatment. This is unlike the synthesised hydrazone compounds, which all theoretically can be administered orally, therefore moving towards better treatment and management of T.b.r infection.
HD4 and HD5 demonstrate lower trypanocidal activity than HD1 and HD6, therefore further progression on HD1 especially would allow for potentially more effective trypanocidal compounds, represented by lower IC50 values upon testing. Furthermore, new compounds in other literature with the highest trypanocidal activity, namely 5-nitro-2-furaldehyde and adamantine alkanohydrazine hydrazones and benzoxaboroles, provide a platform for future work and development of structural analogues of hydrazones to promote trypanocidal activity.
To confirm that the novel compounds synthesised are in fact effective against T.b.r, further testing is required in a controlled level 3 laboratory where the pathogen can be stored safely. Furthermore, no MTT assay or alternative screening test demonstrating the activity against mammalian cell lines was performed, and therefore this would be an area for development to ensure that the drug is effective and safe for potential clinical trials. This could be carried out with use of Hep G2 cells for example, which are human liver carcinoma cells.
Furthermore, research regarding drugs that are able to cross the blood brain barrier would allow for an alternative late-stage treatment to Melarsoprol, and therefore reduce mortality rates should the new compound be less toxic. Pharmaceutical development is desperately required for T.b.r infection to move towards better disease management and possible eradication in the future. Three alternative novel compounds have been designed and are described below.
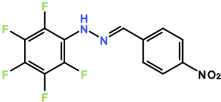 Compound 1: Reacting PFH with 4-nitrobenzaldehyde. Reactive nitrogen species have desirable biologic properties. 4-nitrobenzaldehyde contains an electron withdrawing group, more likely for the compound to react with nucleophiles and therefore form a hydrazone with PFH.
Compound 1: Reacting PFH with 4-nitrobenzaldehyde. Reactive nitrogen species have desirable biologic properties. 4-nitrobenzaldehyde contains an electron withdrawing group, more likely for the compound to react with nucleophiles and therefore form a hydrazone with PFH.
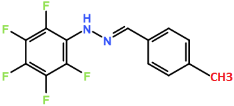
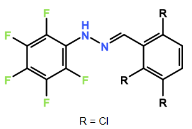 Compound 2: Altering the position of the chlorine ions to the second, fifth or sixth carbon in the aromatic ring may increase trypanocidal activity. R groups shown in figure 4 demonstrate positioning of chlorines.
Compound 2: Altering the position of the chlorine ions to the second, fifth or sixth carbon in the aromatic ring may increase trypanocidal activity. R groups shown in figure 4 demonstrate positioning of chlorines.
 Compound 3: The introduction of a methyl group on one of the aromatic carbons may allow for the compound to become more hydrophobic, which has been shown to promote trypanocidal activity.
Compound 3: The introduction of a methyl group on one of the aromatic carbons may allow for the compound to become more hydrophobic, which has been shown to promote trypanocidal activity.
All compounds proposed abide by Lipinski’s rule of 5 and therefore possibly could be administered orally.
In addition, other factors that may account for future work include altering the concentrations of reactants when synthesising and changing the time exposed to the agent when testing in vitro. These altered parameters may show higher or lower trypanocidal activity, the former potentially moving towards a new therapeutic for HAT. Other than pharmaceutic development, additional research regarding how the therapeutic agent induces a biological response in the human host is another area of research in development. Furthermore, investigations into new drug targets including lipid cell walls or receptors of trypanosomes are underway.
The quality of treatment for T.b.r is limited and with poor efficacy, exemplifying the need for new pharmaceutical development. Current treatment Suramin, Melarsoprol, and co-administration with eflourthrine and nifurtimox are all associated with toxicity and high mortality rates. The aims and objectives of this research project included the synthesis of 4 novel hydrazone compounds to assess their trypanocidal activity in vitro against T.b.b, and hence potentially against T.b.r. The best IC50 values obtained were from compounds HD1, with a value of 9.29 µM, and HD6, with a value of 16.18 µM. Despite these being higher than other compounds in development, the results remain promising and have the potential to aid in the development of an alternative therapy for T.b.r infection. Other hydrazone compounds have demonstrated anti-parasitic properties amongst other properties deemed biologically desirable, therefore provide a good platform for further research and drug design. Three alternative structures have been designed as examples of compounds that may demonstrate trypanocidal activity, although many alternative analogues of hydrazones may demonstrate similar or better results. Further research and drug development appears to be the natural progression for the future of better management and treatment of T.b.r infection. Results demonstrated from hydrazone compounds and derivatives are indeed promising, and further development of these compounds has the potential to massively aid in combatting T.b.r infection.
References
BABOKHOV et al. (2013) A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathology Global Health, 107 (5), pp.242-252.
BART, J.M. et al. (2015) Localization of serum resistanceâ€associated protein in Trypanosoma brucei rhodesiense and transgenic Trypanosoma brucei brucei. Cellular pathology, 17 (10), pp. 1523-1535.
CLAYDEN, J., GREEVES, N. & WARREN, S. (2012) Organic chemistry. 2nd ed. Oxford: Oxford University Press.
FOSCOLOS, A.S. et al. (2016) New hydrazones of 5-nitro-2-furaldehyde with adamantanealkanohydrazines: synthesis and in vitro trypanocidal activity. The Royal Society of Chemistry, 7, pp. 1229-1236.
FRANCO, J.R. et al. (2014) Epidemiology of human African trypanosomiasis. Journal of Clinical Epidemiology, 6, pp. 257-275.
GILBERT, I. H. (2013) Target-based drug discovery for human African trypanosomiasis: selection of molecular target and chemical matter. Parasitology, 141 (1), pp. 28-36.
JACOBS, R.T. et al. (2011) SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 Human African Trypanosomiasis. PLoS Neglected Tropical Diseases, 5 (6).
JONES, A.J., KALSER, M. & AVERY, V.M. (2015) Identification and characterisation of FTY720 for the treatment of Human African Trypanosomiasis. Antimicrobial Agents and Chemotherapy, 60 (3), pp. 1859-61.
KAJAL, A. et al. (2014) Therapeutic Potential of hydrazones as Anti-Inflammatory Agents. International Journal of Medicinal Chemistry.
KATO, C.D. et al. (2015) Interleukin (IL)-6 and IL-10 Are Up Regulated in Late Stage Trypanosoma brucei rhodesiense Sleeping Sickness. PLoS Neglected Tropical Diseases, 9 (6).
KENNEDY, P.G.E. (2013) An alternative form of Melarsoprol in sleeping sickness. Trends in parasitology, 28 (8), pp. 307-310.
KENNEDY, P.G.E. (2013) Clinical features, diagnosis and treatment of human African trypanosomiasis (sleeping sickness). The Lancet Neurology, 12, pp. 186-194.
KOOL, E.T., PARK, D. & CRISALLI, P. (2013) Fast Hydrazone Reactants: electronic and Acid/Base Effects Strongly Influence Rate at Biological pH. Journal of the American Chemical Society, 135 (47), pp. 17663-17666.
LAMOUR, S.D. et al. (2015) Discovery of Infection Associated Metabolic Markers in Human African Trypanosomiasis. PLOS Neglected Tropical Diseases, 9 (10).
LANGOUSIS, G. & HILL, K.L. (2014) Motility and more: the flagellum of Trypanosoma brucei. Nature Reviews Microbiology, 12, pp. 508-518.
LEESON, P. (2012) Drug discovery: Chemical beauty contest. Nature, 481 (7382), pp. 455-456.
LIU, T.C. et al. (2014) The unique chemistry of benzoxaboroles: current and emerging applications in biotechnology and therapeutic treatments. Bioorganic & Medicinal Chemistry, 22 (16), pp. 4462-4473.
MORGAN, H. P et al. (2010) The Trypanocidal Drug Suramin and other Trypan Blue Mimetics Are inhibitors of Pyruvate Kinases and Bind to the Adenosine site. Journal of Biological Chemistry, 286 (36), pp. 31232 – 31240.
PALMER, M. J. & WELLS, T. N. C. (2012) Neglected Diseases and Drug Discovery. Cambridge: The Royal Society of Chemistry.
PEPIN, J. (2014) Trypanosoma brucei gambiense and Trypanosoma brucei rhodeisense (African Trypanosomiasis). Journal of infectious diseases and antimicrobial agents, Available from: http://www.antimicrobe.org/b54.asp [Accessed 07/11/16].
RABER G. et al. (2013) Determination of the trypanocidal drug Melarsoprol and its conversion products in biological fluids with HPLC-ICPMS/ESMS. Talanta, 116, pp. 876-881.
STEVERDING, D. (2010) The development of drugs for the treatment of sleeping sickness: a historical review. Parasites and Vectors, pp. 3-15.
STIJLEMANS, B. et al. (2016) Immune Evasion Strategies of Trypanosoma brucei within the Mammalian Host: Progression to Pathogenicity. Frontiers in Immunology, 7 (233).
TORREELE, E. et al. (2010) Fexinidazole – A new oral Nitroimidazole drug candidate entering clinical development for the treatment of Sleeping Sickness. PLoS Neglected Tropical Diseases, 4 (12).
VERMA, G. et al. (2014) A review exploring biological activities of hydrazones. Journal of Pharmacology & Bio allied Sciences, 6, pp. 69-80.
NCBI PUBCHEM(2017) Suramin
Appendix 1

Figure 2: General mechanism of hydrazone formation. PFH (hydrazine) acts as a nucleophile, reactants with carbonyl group act as electrophiles.
Note: R1 in this generic formula should be substituted with relevant benzene rings shown in figure 1 (reactants A-D) to demonstrate the complete reaction mechanism.