L. Pisonis Nut Oil Extraction
The search for new crops to provide vegetable oils for use in the industry and also for human consumption is growing increasingly over the past two decades. A rapid search on Web of Science„¢ platform present more than 19200 results related to “nuts”, more than 7600 documents are related to “edible oils”, and more than 22000 results associated with “vegetable oil”. Additionally, the published items regarding vegetable oil increased from 282 in 1996, to more than 1930 in 2016 proving the growing interest by the scientific community in such field of study [1].
Several Brazilian seeds/nuts species, especially from the Amazonian area have been studied during the last years and most of them were shown to present bioactive compounds and also potential as functional foods [2]-[7]. Nevertheless, some other species are not well-known or were not summited to any further study to elucidate other characteristics.
Lecythis pisonis Camb., a Lecythydaceae tree that grows in Brazil, Colombia, Venezuela and in the Guyanas, commonly known as ”sapucaia” provide nuts particularly similar with that of Brazil nut (Bertholletia excelsa). The delicious edible kernels of sapucaia present a characteristic sweet flavor, being considered more digestible than Brazil nut [8]. In addition to the economic and ecological importance of sapucaia, often used to shade cocoa plantations in Brazilian agroforestry systems [9], the nuts from L. pisonis are a valuable source of macro and micronutrients, essential amino acids, minerals such as P, K, S, Mg, Ca, Mn, Ba, Zn, Fe, Sr, Cu, B, Al, and Cl [3], [10], and also fibers [11]. Furthermore, the sapucaia nuts constitute a relevant source of lipids, about 51-64% [5], [8], [10], [11], in which there is a predominance of the linoleic acid (essential fatty acid).
Although sapucaia can be considered a potential crop to provide raw material for the production of vegetable oil, the available papers that studied the nuts have mostly focused on the mineral and fatty acid profile [3], [5], [10], [12], [13], and the latest reports are limited to some agronomic, microbial and pharmacological aspects [9], [14]-[17].
Vegetable oils are widely utilized in many applications such as food, cosmetic, pharmaceutics, and biofuel industries. Consequently, stability tests, rheological essays, phase transitions and other properties of the oils should be well characterized to optimize processing conditions and energy inputs [18], [19].
No reference has been found regarding the total phenolic compounds, antioxidant activity, thermal and rheological characterization of sapucaia nut oil. Therefore, the aim of this work was to study the sapucaia nut oils extracted by Soxhlet and Bligh & Dyer means and evaluated for their fatty acid composition, rheological behavior, and thermal properties by using DSC and TG/TGA analysis, total phenolic content, antioxidant properties and oxidative stability by Rancimat in addition to Attenuated Total Reflectance-Fourier transform infrared spectroscopy (ATR-FTIR).
2.1 Chemicals
Other reagents used in the experiments were of analytical grade. The aqueous solutions were prepared using ultrapure water.
2.2 Samples
Lecythis pisonis nuts were harvest from a crop area located in the city of Araguanã, Maranhão State, Brazil. The nuts (Fig. 1) were dried in an air-circulating oven (Soc. Fabbe, Brazil) at 40 °C for 24 h. Nutshells were removed manually using a stainless steel knife and then submitted to freeze-drying (Liotop L101, Liobras, Brazil) to remove the residual moisture. Prior to the oil extraction, the nut samples were crushed for 30 s with the aid of a stainless steel knife grinder (MA630/1 Marconi Ltda., Brazil).
2.3 Extraction of nuts oils
The oil content of L. pisonis nut samples was extracted by the Bligh & Dyer method described in AOCS Ba 3-38 method, and also using n-hexane with Soxhlet apparatus (Vidrolabor®, Labor Quimi, Brazil) according to American Oil and Chemical Society Official Method (1997). Oils were named LP1 and LP2, respectively.
After the oil extraction, solvents were removed at 43 °C under reduced pressure using a rotary evaporator (Model 801, Fisatom Ltda., Brazil). Samples were dried at 45 °C in an air circulating oven (Solab, Brazil) and then flushed with gaseous N2 before storage. The oils were kept in an amber glass and stored at -10 °C until further analysis.
2.4 Analytical determinations
2.4.1 Water content and apparent pH
Water content was measured by volumetric Karl Fischer titration using a titration system (V30 Mettler-Toledo, Switzerland) [21].
The determination of the pH was performed according to Pena Muniz et al. (2015), as recommended by the Brazilian National Agency for Sanitary Surveillance without previous dilution of the oil, with the aid of a digital pH meter (Model PG1800 Gehaka) at 25 ± 2 °C. The results represent the mean of three measurements.
2.4.2 Measurement of fatty acid composition
Fatty acid composition of fresh L. pisonis oils was investigated according to AOCS Official Method Ce 1a-13 [20] by using a GC-MS system (GC-2010) coupled to a mass spectrometer (GCMS-TQ8030) and automatic injection system by headspace (Shimadzu, Japan). Oil samples solubilized with hexane were methylated as described by AOCS Ce 2-66 and then injected into a capillary HP88 column (60 m Ã- 0.250 mm i.d., 0.2 μm film thickness, Agilent Technologies) using helium as carrier gas (0.64 mL/min). Oven temperature was programmed to be 175 °C (isothermal) and the detector was held at 250 °C. An aliquot of 1 μL of the sample was injected using the split injection mode (1:100). Fatty acid composition was expressed as the percentage of the total peak area of all the fatty acids in the oil sample.
2.4.3 Rheological studies
Rheological measurements of oils were performed according to Teixeira et al. (2016), by using a Haake Mars II (Thermo Electron GmbH, Germany) rheometer connected to a thermostatic bath (Haake K15), a thermo-circulator water unit (Haake DC5B3) and a Peltier temperature control (Haake UTM Controller). A sensor cone-plate (C60/2 °Ti L) with a diameter of 60 mm and a cone angle of 2 ° was used. Samples remained in the rheometer for 5 min prior to analysis, and sampling was carried out in duplicate. The following measurements were performed: i) flow curve (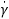 = 0.01-500 s-1, t = 300 s); ii) dynamic stress sweep (Ï„ = 0.01-100 Pa, ω = 1 Hz) and subsequent dynamic frequency sweep (ω = 0.1-100 Hz, Ï„ = 1.0 Pa); and iii) temperature sweep (T = 10-60 °C, t = 1440 s, 2 °C/min and 60-10 °C, t = 600 s, 5 °C/min; ω = 1 Hz, Ï„ = 1.0 Pa). Temperature sweeps were performed at a constant tension of 0.5% in the Linear Viscoelastic Region (LVR). The samples were covered by using a sample hood in order to avoid interference of the ambient temperature.
= 0.01-500 s-1, t = 300 s); ii) dynamic stress sweep (Ï„ = 0.01-100 Pa, ω = 1 Hz) and subsequent dynamic frequency sweep (ω = 0.1-100 Hz, Ï„ = 1.0 Pa); and iii) temperature sweep (T = 10-60 °C, t = 1440 s, 2 °C/min and 60-10 °C, t = 600 s, 5 °C/min; ω = 1 Hz, Ï„ = 1.0 Pa). Temperature sweeps were performed at a constant tension of 0.5% in the Linear Viscoelastic Region (LVR). The samples were covered by using a sample hood in order to avoid interference of the ambient temperature.
The coefficient of determination (R2) and chi-square (χ2) were chosen to evaluate the fit of the Ostwald-de Waele (Eq. (1)) and Herschel-Bulkley (Eq. (2)) models to the experimentally obtained flow curves:
|
– Ostwald-de Waele (OW) |
(1) |
|
|
– Herschel-Bulkley (HB) |
(2) |
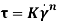
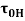 +
+ 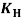 (
(where Ï„ is the shear stress (Pa), 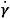 is the shear rate (s-1), Ï„0H is the HB yield stress (Pa), K and KH are the consistency index (Pa·sn), and n and nH are the flow behavior indexes (dimensionless).
is the shear rate (s-1), Ï„0H is the HB yield stress (Pa), K and KH are the consistency index (Pa·sn), and n and nH are the flow behavior indexes (dimensionless).
The effect of the temperature on apparent viscosity was evaluated according to an Arrhenius type equation (Eq. (3)), using a shear rate of 53.4 s-1.
|
|
(3) |
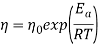
where 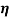 is the apparent viscosity at a specific shear rate,
is the apparent viscosity at a specific shear rate, 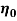 is the preexponential factor,
is the preexponential factor, 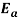 is the activation energy (J.mol-1),
is the activation energy (J.mol-1), 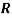 is the gas constant (8.31 J. K-1. mol-1) and
is the gas constant (8.31 J. K-1. mol-1) and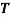 is the absolute temperature (K).
is the absolute temperature (K).
2.4.4 Oxidative Stability Index
The oxidative stability index (OSI) was estimated in a Metrohm Rancimat model 743 (Herisau, Switzerland), following the American Oil Chemists’ Society Official Method Cd 12b-92 [20]. Briefly, increasing water conductivities were continually measured while air (20 L/h) was bubbled into the oil (3 ± 0.1 g) heated to 110 °C and their volatile compounds were collected in water. The time taken to reach the conductivity inflection time was recorded. IP was registered by Rancimat 743 PC Software 1.1.
2.4.5 Infrared spectroscopy
Fourier transform infrared spectroscopy (FTIR) analyses were determined by using a Vertex-70 spectrometer (Bruker, USA) with an attenuated total reflectance (ATR) accessory, at 25 °C. The oil samples were placed uniformly in the ATR crystal (1.5 mm active area). The FTIR spectra were recorded in the wavenumber range of 4000 to 600 cmˆ’1 with a spectral resolution of 4 cmˆ’1 [23].
2.4.6 TG-TGA Thermal decomposition
TGA analyses were performed in TGA 4000 equipment (PerkinElmer Inc. Waltham, USA). Oil sample (~10 mg) was placed into the platinum pan and then placed in a furnace. The sample was heated from 30 to 750 °C (10 °C/min) in the air atmosphere (70 mL/min flow rate) or under a nitrogen atmosphere. Data on the weight changes of oil samples were obtained from the Pyris„¢ software. TG and derivative thermogravimetric (DTG) curves were further analyzed by using Origin 8.6 software (OriginLab, Massachusetts, USA). The thermal stability was measured from the extrapolated onset temperature of the first step of thermal decomposition from respective TG curves, by using the beginning and the peak temperature of the respective DTG peak, as the temperature limits of the data analysis software of the instrument.
2.4.7 DSC Analysis
For DSC analysis, a DSC 8500 (PerkinElmer Inc. Waltham, USA) equipped with Pyris„¢ software was used. Nitrogen (99.99% purity, White Martins, Brazil) was the purge gas (20 mL/min). The DSC instrument was previously calibrated with Indium (m.p. 156.6 °C, ΔH = 28.45 J/g). The sample was weighed into an aluminum pan and sealed. An empty hermetically sealed aluminum sample pan was used as the reference. In order to reduce temperature gradients, the sample mass was kept small (~ 9.0 mg). The methodology applied was adapted from Zhang et al. (2013).
For cooling and melting curves, the samples were first rapidly heated to 80 °C (30 °C/min) from room temperature and held for 10 min. Then, the samples were cooled to ˆ’80 °C (10 °C /min) and held for 10 min to make them fully crystallized, then heated to 80 °C (5 °C /min).
To study the effect of the cooling rate on crystallization, samples were rapidly heated to 80 °C (30 °C/min) from room temperature to erase the crystallization memory, and held for 5 min, then the samples were cooled to ˆ’80 °C at the cooling rates of 2 °C/min, 5 °C/min and 10 °C/min, respectively. The cooling thermogram was recorded.
For investigating melting profile of L. pisonis oils under isothermal crystallization, the samples were heated at 80 °C (30 °C/min) for 5 min and then cooled (100 °C/min) to a preset temperature (ˆ’10 °C, 0 °C, 10 °C, 20 °C), and hold at that temperature for 10 min for crystallization, then heated to 80 °C (5 °C/min). The melting thermogram was recorded.
Pyris„¢ manager software was used to calculate the parameters of crystallization and melting, enthalpies, and the solid fat content (SFC) which was estimated based on the integrated area under the melting curve [25]. To measure the cloud point of the oils, the cooling curves at the rate of 2 °C/min were used. The cloud point was the onset temperature of the initial small exothermic peak on these cooling curves [26].
2.4.8 Total phenolic compounds
Contents of the total phenolic compounds (TPC) were determined using the Folin-Ciocalteu reagent assay in methanolic extracts of vegetable oils [27]using microplates technique [28],with gallic acid as a standard for the calibration curve. The absorbance at 720 nm was measured using a spectrophotometer (Tecan Nanoquant Infinite® M200, Tecan Trading AG, Switzerland) after reaction with the Folin-Ciocalteu reagent in the alkaline medium stand in the dark for 1 h. Contents of the TPC were expressed in mg of gallic acid equivalents (GAE)/100 g of oil.
2.4.9 Antioxidant essays of the oil extracts
The free radical scavenging evaluated by the DPPH assay was determined in triplicate using the method proposed byBrand-Williams et al., (1995). The absorbance at a wavelength of 517 nm was measured using a spectrophotometer (Tecan Nanoquant Infinite® M200, Tecan Trading AG, Switzerland). ABTS scavenging activity of oil extracts was determined in triplicate using the method described by Re et al., (1999). The absorbance at 734 nm was measured. The total antioxidant potential of the oil extracts was performed using the ferric reducing antioxidant power (FRAP) assay [31]. Measurements were performed using the spectrophotometer at 593 nm. The absorbance of the samples was compared to a standard curve (100-1000 µmol/L) and results expressed in mmoL Trolox equivalent per g of oil [mmol TE/g]. All the essays had minor changes as proposed by Zielinski et al., (2016). The determinations were performed in triplicate.
2.5 Data analysis
Origin 8.6 software (OriginLab, Massachusetts, USA) was employed to data treatment and graphs. Statistica 10.0 (Statsoft, Brazil) was used for statistical analysis, including means and standard deviations (SD). The experiments were carried out in triplicate.
3.1 Water content and apparent pH
As expected for this type of raw material, sapucaia (Lecythis pisonis) oil (SO) samples presented very low water content, to be specific 0.077 ± 0.006 %, and 0.097 ± 0.006 % for LP1 and LP2, respectively. Regarding the apparent pH of SO, LP1 presented 5.720 ± 0.036 and LP2 showed a pH value of 5.353 ± 0.040.
3.2 Phenolic compounds and antioxidant properties
Concerning the content of phenolic compounds in SO, LP1 presented higher TPC content than LP2(Table 1). In this regard, SO present greater TPC content (1.418 mg GAE/100 g, average value) than almond oil (0.95 mg GAE/100 g), however, Brazil nut has twice the content of TPC (3.64 mg GAE/100 g) than sapucaia, and macadamia presents fifteen times more TPC (22.5 mg GAE/100 g) [33].
In general terms, the sample LP1 obtained by a cold extraction process showed better antioxidant characteristics than LP2. Natural antioxidants that might prevent biological systems from reactive oxygen species are generally present in vegetable oils as phenolic compounds, phytosterols, tocopherols (Vitamin E) and carotenoid compounds [34]. The results found by in vitro antioxidant assays present some variations that might be assigned to the different mechanisms in the analysis (Table 1). These assays are grouped in the electron-transfer category and there is some specificity among them. DPPH and ABTS radical cations have the same mechanism and present characteristic color which can be monitored by a spectrophotometer when the oil extract is mixed with a reactant solution and the reduced form of the radical shows a loss of color as a result of the donation of a hydrogen atom. Similarly, in the presence of antioxidants compounds, FRAP is characterized by electron transfer ability, that results in the reduction of iron ions [35]
3.3 Fatty acids profile
Fourteen different fatty acids (FA) were identified in the studied SO (Table 2). The oils were composed predominantly of unsaturated fatty acids (UFA) (61-73%), in which 39.08-45.66% were monounsaturated (MUFA), and 21.95-27.63% were comprised of polyunsaturated (PUFA), such as linoleic and α-linolenic acids, which have been proved to effectively reduce the risk of cancer, osteoporosis, cardiovascular diseases, and diabetes [36]. Such FA profile is rather common for Brazilian edible nuts [5], [37], which reinforces the idea of consuming more nuts rich in fatty acids beneficial to human health, since the main commercial source of PUFA, such as ω-3 and ω-6 has been fish and fish fat [36].
Among the MUFAs, oleic was the major FA found in SO, ranging from 35.54-44.28% for LP2, and LP1, respectively. Important to note that oleic acid is the primary ω-9 FA in the human diet, and it is the predominant MUFA in many oil nuts, totalizing, for example 38.50% in Brazil nut (Bertholletia excelsa) [22], 65.59% in garampara (Dipteryx lacunifera) [5], 67.62% in tucumã (Astrocaryum vulgare Mart.) and 39.04% in cutia nut (Couepia edulis) [37]. Besides, the SO presented 21.65-27.19% of linoleic acid, an essential FA which presence in reasonable amounts is important for human health [38]. SO also contains two important cis-MUFA, the cis-Vaccenic and cis-11-Eicosenoic in amounts higher than 1.0%.
With regard to the saturated fatty acids (SFA), it is noticed that they represented 25.01-37.25% of total FA, mainly palmitic (14.70-21.38%) and stearic (9.63-11.09%); likewise, other SFA such as myristic, heptadecanoic, and behenic were identified in smaller amounts. Arachidic acid, which is usually present in peanut oil was also found in SO in small quantities and the result is in agreement with Costa and Jorge (2012) who found about 0.22% C20:0 for L. pisonis oil. A similar pattern occurs for Brazil nut, which shows 0.36% arachidic acid, and 14.26% palmitic acid [22].
A good factor to evaluate the quality and digestibility of a vegetable oil can be obtained by the amount and composition of UFA and SFA. A high amount of linoleic acid in comparison to oleic acid (ω-9) represents a better quality of the vegetable oil [5]. In this regard, the relationship between linoleic/oleic acids should be considered. The oils can present diverse values for this relationship depending on the extraction method. According to Table 3, the UFA/SFA ratio, as well as the linoleic/oleic ratio for sapucaia oils are lower to those found by [5] and [10], once their oils showed higher content of linoleic acid (Table 2). When compared to other Brazilian oleaginous nuts such as Brazil nut, garampara [5], and cutia nut [37] the unsaturation relationship values between linoleic and oleic acid were also quite different.
3.4 Oxidative Stability Index
The oxidative stability index (OSI) is considered an important step in evaluating oil quality. The OSI was evaluated during Rancimat analysis until the end point of stability for SO samples and was expressed in hours. Experimental results confirmed that Bligh & Dyer (13.28 ± 0.22 h), which is a cold extraction process, is a technique that causes less damage to the oil in comparison to Soxhlet (7.18 ± 0.50 h) which provided almost one-half of the OSI. The high oleic acid content in SO tends to protect the oil against thermo-oxidation. Costa and Jorge (2012) found 24.89 h of OSI (evaluation at 100 °C) for L. pisonis oil extracted by cold pressing. At the same Rancimat conditions used in this study, similar results were found for Brazil nut oil (8.24 h), hazelnut (8.88 h) and macadamia (7.38 h) [33].
3.5 Thermal decomposition
Thermogravimetric curve (TG) shows the mass loss, and the derivative thermogravimetric curve (DTG) shows the rate of mass loss of SO during thermal decomposition from 30 to 750 °C by TGA (Fig. 2). The values of mass loss are indicated in Table 4. The TG curves indicated that SO were thermally stable up to 303 °C with a mass loss of ~5%. This can be explained by the loss of moisture and volatile compounds in the oils. The oxidative process in vegetable oils is characterized initially by the oxidation forming secondary products (peroxides). The following phase corresponds to the decomposition of MUFA, mainly oleic acid and the polymerization of the substances remaining from the previous phase [39].
Dynamic and inert atmospheres cause different pattern in the TG/DTG profiles. The decomposition and carbonization processes in the air atmosphere occurred in three phases of the curve starting at around 130 °C, achieving about 53-58% mass loss at 405-440 °C, and ending at a temperature range of 611-625 °C, while for nitrogen atmosphere, the process showed two steps, initiating at ~160 °C, reaching 62-75 % mass loss at 426-435 °C, and finishing at 500 °C. DTG curves (Fig. 2) shows more clearly the steps of thermal decomposition. At the higher above-mentioned temperatures, the mass loss reached 100% (no residue remaining). The high UFA content in SO, mainly oleic and linoleic acid are related to be responsible for the occurrence of oxidative degradation reactions [22]. The high values of Ti (onset temperature) showed that SO have high thermal stability, being that the higher is the Ti of decomposition of the oil, the higher is the thermal stability.
The results are similar to that of Brazil nut, which is thermally stable up to 209-220 °C and reaches a maximum mass loss of 97% at 580-602 °C [22], [39]. Differences between the thermal behaviors of SO can be related to the FA compositional differences, and also because of the presence of natural antioxidants which tends to protect the oils against oxidation, thus retarding the degradation [40].
3.6 Thermal behavior for crystallization and melting
3.6.1 Temperature for crystallization and the influence of scanning rate
In order to investigate the melting profile of SO under isothermal crystallization, previous tests were done. In one hand, the melting thermogram (Fig. 3 a, b) revealed that when frozen at -10 °C the oil samples showed endothermic peaks at -6.81 °C (LP1) and at -6.16 °C (LP2), with a enthalpy of 9.28 J/g for LP1 and 7.26 for LP2. On the other hand, it was demonstrated that there was no event afterward 0 °C, revealing that no crystallization occurs after that temperature. Such a feature may confirm the liquid state of the SO at room temperature (25 °C).
During cooling, the oils exhibited two transitions. The thermograms showed that the position of the exotherms is dependent on the cooling rate, while the amount of the exotherms is independent on the cooling rate. In addition, increasing the cooling rate causes the crystallization peak temperature to shift to lower temperatures, the peak height and area increase (Fig. 3 c, d). This behavior may correspond to the differential crystallization of higher melting triacylglycerols (TAGs) firstly and lower melting TAGs secondly. Similar results are described for palm oil fractions [24].
The scanning rate of 2 °C/min reduces the lag in output response from the DSC instrument as well as preserves the minor peaks and reduces the smoothing tendencies, which occurred at a higher scanning rate; however, the first peak is too small when compared to the rate of 5 °C/min, which provided better resolution for the peak analysis in the Pyris„¢ software.
3.6.2 Cooling curve
During cooling (Fig. 4a), SO presented a similar pattern of crystallization showing two distinct peaks (exothermic) in the crystallization behavior, namely peak 1 (PC1) and peak 2 (PC2). PC1 which indicates the change from liquid to solid was found in the temperature of -11.56 (LP1) and -12.51 (LP2), while the PC2 was observed in the temperature range of -59.80 to -73.93 °C, both related to the crystallization of TAGs. The crystallization onset temperature occurs at around -8.0 °C and this process extends over a range of 15-23 °C. The sum of the crystallization enthalpy of the two peaks for the two oils were -22.17 and -32.56 J/g for LP1 and LP2, respectively (Table 5). Heat is released during the phase transition of oil from liquid form to solid form; for this reason, values for crystallization enthalpy were negative [41].
Different endothermic and exothermic peaks are exhibited according to different contents of saturated or unsaturated TAG and FA present in the oils [42]. Smaller or shoulder peak are also correlated to the differing type of TAG. In addition, an inseparable shoulder peak which melts at the same temperature range comes from the complex nature of the TAG [43].
3.6.3 Heating curve
During melting, SO were found to be completely melted at 8.28 °C and 6.29 °C (LP1 and LP2, respectively) when heated at 5 °C/min (Fig. 4b). Moreover, LP1 showed a single major shoulder before the maximum melting peaks (PM) at -18.42 °C, while LP2 showed a major shoulder before PM (-21.47 °C) and an additional one, after the major peak at -6.61 °C. The melting curve of SO began at -25.77 °C (LP1), and -29.87 °C (LP2) and the major peak was observed in a temperature region varying between -7.99 to -10.13 °C, ending the events around -1.09 to -1.68 °C. The process comprises a melting range of 24-28 °C, with a melting enthalpy of 59.34 (LP1) and 64.76J/g (LP2) (Table 5).
During heating treatment of oil, multiple endothermic regions are correlated to the separate melting events of the TAG. Furthermore, the composition and polymorphism of the oils can cause some overlapping effects, resulting in different shapes of the endothermic peaks [18]. The phase transitions are principally determined by the degree of heterogeneity in the composition of the oils, and include breaking/formation of hydrogen bonds, being a result of structural rearrangements in the chains of glycerides. [44].
3.6.4 Cloud point
The cloud point is referred as the temperature at which oil begins to cloud as a result as a result of the first stage of crystallization under controlled cooling, and it is related to the oils’ degree of unsaturation. The higher is the unsaturation of the oil, the lower the cloud point [26]. The cooling curves of sapucaia oils showed an initial small exothermic peak at -6.87 °C (LP1) and -7.00 °C (LP2). This peak corresponded to the first stage of crystallization and cloud point corresponded to the onset of crystallization. A second large exothermic peak at ˆ’49.95 °C (LP1) and -49.96 °C (LP2) with further cooling indicated the oil underwent an additional crystallization step. The same behavior was found for hazelnut oils, which presented the first small exothermic peak at -12.4 °C and a large one at -31.5 °C [26]. The lower cloud point of sapucaia oils was also a result of the lower content of SFA compared to the UFA.
3.6.5 Solid Fat Content
The solid fat content (SFC) is considered an important physical property of lipids because it expresses their physical, sensorial, technological and protecting/release properties, and it affects physical properties such as consistency, spreadability, and stability [45]. According to the melting thermograms (Fig. 4b), SO presented no changes in SFC previously to -40 °C, so then the data analysis was performed in the range of -40 to 20 °C (melting peak region). The SFC showed less than 2.5% decrease between -40 to -30 °C for the two oils, and started to drop rapidly at around -20 up to 0 °C; then slowed down from 6 to 10 °C until there was no solid fat remaining (Fig. 5). The SFC is associated with the FA profile. Oils with higher content of UFA are easier to melt compared to ones with higher SFA [41], [45].
3.7 Rheology properties
3.7.1 Steady state rheology
The results for rheological behavior of SO shows that the apparent viscosity (η) tends to decrease with increasing temperature (T) and also with increasing the shear rate (γ), indicating that the flow of the sapucaia oils is pseudoplastic (n > 1) (Table 6, 7 and Fig. 6). On the one hand, it is usual for oils to exhibit a dependency of T; on the other hand, a non-dependency of higher γ is observed. At 50 °C, the η is almost constant throughout the entire range tested, with minimum influence of the γ. Furthermore, 2.0 s-1wasthe maximum value of γ which induced major changes in η (see detail in Fig. 6, b and c). According to Gila et al. (2015), a greater amount of FAs such as C18:1 and C18:2 as the major components of the oils appear to make a great contribution to the flow behavior of oils.
The experimental data fitted by Ostwald-de Waele (OW) and Herschel-Bulkley (HB) models (Fig. 6a, Table 6) showed a good fit (R2 > 0.999; χ2 < 0.012). In agreement with the η, as T increases, the K and KH values decrease (Fig. 6e and Table 6). Similar observation for many edible oils such as coconut, sunflower, canola, corn [47] and olive oils [48] was reported. The OW model showed that the oils were non-Newtonian (n < 1) over the T range tested. However, the HB model revealed that sample LP2 behaved as a dilatant fluid (nH > 1) at 30 and 40 °C (Fig. 6f). The HB model is able to determine yield stress (Ï„0H), at which a material begins to deform plastically without returning to its original shape when the applied stress is removed. SO have small values for Ï„0H which also tends to decrease with the increase of T.
Fig. 7 shows the non-linear and linear relationship of η vs T, and ln η vs. 1/T, respectively, obtained from Arrhenius equation (Eq. 3) and fitted to experimental data (Table 7), proving the η dependence of T. This could be due to the energy obtained to overcome the resistance to flow, which may be due to the attractive forces among the oil molecules. This behavior is common for vegetable oils such as coconut oil, crude palm kernel oil, olive oil, sesame oil, soy oil, sunflower oil [49].
In vegetable oils, Ea is related to the degree of PUFA. Usually, a high poly-unsaturation (linoleic and linolenic acid content) would lower and high oleic content in the FA chain would increase the Ea for oxidation [50]. The greater the activation energy (Ea) found indicates the greater sensitivity of viscosity of the sample when the temperature is modified. In addition, the higher the Ea, the higher is the stability to oxidation. The Ea for LP1 was slightly higher (31.89 kJ/mol) than LP2(29.66 kJ/mol) being, therefore, the sample with greater η variation when changing T.
3.7.2 Oscillatory rheology
Viscoelasticity is usually applied to provide valuable information of oils relating its behaviors with the composition and other physical properties. The storage modulus G’ is correlated to elasticity (solid-like), whereas the loss modulus G” is associated to viscosity (liquid-like). The complex modulus |G*| reveals the overall structure of the system, while the phase angle δ suggests whether a fat system is more solid-like or liquid-like [51].
As revealed by Fig. 8a, the gradual decrease in the |G*| during heating followed by the opposite effect during cooling could reveal the destruction of a primary crystal network (melting effect) which was crystallized again when the oil was cooled, once |G*| returned to the initial values, thus to strengthen the possible crystal network that was about to form again close to 10 °C [51]. In all samples, the elastic (G’) and viscous (G”) components increased with the applied frequency over the range from 0.1 to 10 Hz, with G” always being higher than G’ (Fig. 8 b, and c). Therefore, the samples were all viscoelastic fluids with a predominant viscous component. Similar behavior is reported for olive-pomace oils [48].
3.8 ATR-FTIR analysis of the sapucaia oils
In Fig. 9 each peak corresponds to functional groups responsible for IR absorption; meanwhile, the intensities of each peak are associated with the concentration of functional groups present in both oils. To the naked eye, the entire range of spectra looks very similar for the SO. There are no considerable differences between their spectral features apart from slight changes in the absorbances of some bands as well as some shifts in the exact position of the bands. In general terms, the spectra displayed the characteristic peaks of functional groups common to ester samples [44], [52]. Some differences were only observed at the absorption bands around 2955, 1261, 804 and 756 cm-1 (Table 8).
Peaks at region 3008 cm-1 attributed to cis -C=CH vibration and at 1653 cm-1 caused by vibration of cis C=C were present in both samples. This peak is also correlated with the presence of UFAs [44], [52], [53].
The strong bands at around 2922 and 2854 cm-1 can be ascribed to the asymmetrical and symmetrical C-H stretching vibrations of CH2 groups, which are also present in macaúba palm (Acrocomia aculeata) oils [44]. The triglyceride, which is a major component in edible oils and fats, was dominant in the spectra. The major peaks that represent triglyceride functional groups could be observed around 2922 cmˆ’1, 2854 cmˆ’1, 1745 cmˆ’1, 1464 cmˆ’1, 1161 cmˆ’1, and 723 cmˆ’1 [53].
A small shoulder at 1711 cmˆ’1, assigned to free FAs was also detected. The band at 1464 cmˆ’1 is the scissoring band of the bending vibration of the methylene group. A band at 1417 cmˆ’1 is attributed to rocking vibrations of CH bonds of cis-disubstituted olefins [53]. Likewise, a band, at 1377 cmˆ’1 could be due to symmetrical bending vibration of methyl groups. The band at around 1161 cm-1 may be assigned to the stretching of the C-O bonds of aliphatic esters or CH2 bending vibrations [54]. In addition, the peak at 1161 cmˆ’1 and its shoulder at 1238 cmˆ’1 are considered as fingerprints of C-O stretching in long-chain fatty acids [24]. The sapucaia oils presented a small peak in the region of 968 cm-1, which is assigned to trans UFAs. Finally, the band at 722 cmˆ’1 is attributed to (CH2)n rocking [53].
These differences between the FTIR spectra of LP1 and LP2 confirm that the extraction method caused some alterations on the chemical profile of the oils. This finding also suggests that the oil composition can affect the exact position of the bands and also affects the shifts in the IR spectra.