Modification of the 4-Quinolone Scaffold
Discovery:
Quinolones are antibacterial agents that are of major importance in the antibacterial field as the can act as the model antibiotic. This is due to their broad range of activity, the high potency and good bioavailability with both intravenous and oral administration possible. This is coupled with high serum levels and a distribution within tissues that specifies concentration levels and results in, theoretically, few occurrences of unwanted side effects.1 The timeline for the development of this class of antibacterial agents begins with the isolation of the bactericidal naphthyridine, nalidixic acid, in the 1960s by George Lesher as the first synthetic quinolone antibiotic. Nalidixic acid is illustrated in Figure 1 below and is a by-product that was isolated from a chloroquine synthesis.2
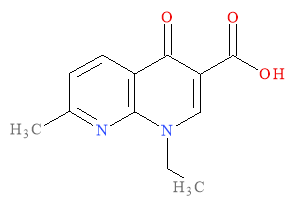
The clinical use for naldixic acid was to treat urinary tract infections (UTIs) caused by gram negative organisms. The successive generations of quinolones had activity against both gram negative and gram positive bacteria as well as anaerobic bacteria. This development lead to fluoroquinolones which are latest in quinolone antimicrobials. The clinical uses for the quinolones today include respiratory tract infections, bacterial meningitis and gastrointestinal infections as well as the historical use of treating UTIs. The development of fluoroquinolones resulted in a more extended spectrum of activity and improved pharmacokinetics then the first generation of quinolones.3,4
Structure:
The general quinolone class of molecules is comprised of 4-quinolone and 1,8-napthyridine ring structures. The naphthyridine ring structures differ slightly from the 4-quinolone core due to the presence of two nitrogen atoms in the rings of the molecule (Figure 2). The substituents R5, R6, R7 and R1 were added to improve the activity of the quinolone core before the development of highly potent fluoroquinolones.1
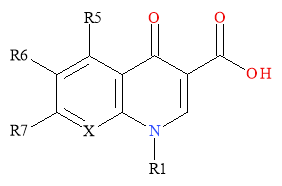
Figure 2: General Structure of 4-Quinolones
The first fluoroquinolone to be developed was Flumequine; illustrated in Figure 3. It had a fluoro-group at the 6 position and was the first compound to show that modifications of the quinolone core could results in improved activity against the gram-positive bacteria that nalidixic acid had no effect on.1
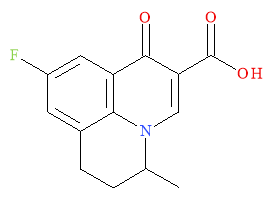
Figure 3: The first fluoroquinolone- Flumequine
Mechanism of Action:
Inhibition of bacterial DNA gyrase (topoisomerase II) and topoisomerase IV is achieved by quinolones. These enzymes play a vital role in the uncoiling of DNA. DNA gyrase acts as the target in gram negative microbes and topoisomerase IV as the target in gram positive microbes for quinolone activity. The widely-accepted mechanism of action is that quinolones bind to complexes, formed between DNA and one of the enzymes, to generate a Quinolone-DNA-Enzyme complex that inhibits DNA replication. The binding of quinolones and topoisomerase is enabled by a water-metal ion bridge. The inhibition is bacteriostatic as replication is reversible. Eventually apoptosis occurs due to the fragmentation of the DNA ends of the complex. This results in bactericidal inhibition. The most common form of resistance to the quinolones is due to specific mutations in the topoisomerase II and IV that interfere with the water-metal ion bridge interaction.2,3
Development:
Quinolones are grouped into generations depending the activity of the molecules. The first generation showed activity against gram negative bacteria that caused UTIs. The second generation showed enhanced activity against gram negative bacteria and improved activity against gram positive. This enabled the list of conditions that quinolones could treat to expand. This generation displayed improved pharmacokinetics; due to the use of a C7-piperdinyl substituent. The third generation provided improvement in efficacy in inhibiting gram positive and anaerobic pathogens. 3,5 The fourth generation of drugs observed dramatically increased activity against DNA gyrase and gram positive microbes, improved pharmacokinetics and pharmacodynamics. The major changes were the addition the fluoro-group at the C-6 position and a ring substituent at C-7. Norfloxacin (1), second generation, was the first broad spectrum quinolone with ciprofloxacin (2) the first quinolone to have activity observed beyond the treatment of UTIs.2 Currently, Garenoxacin (3), fourth generation, is of interest due to its distinct carbon-carbon bond at position C7 and its broad spectrum of activity.3
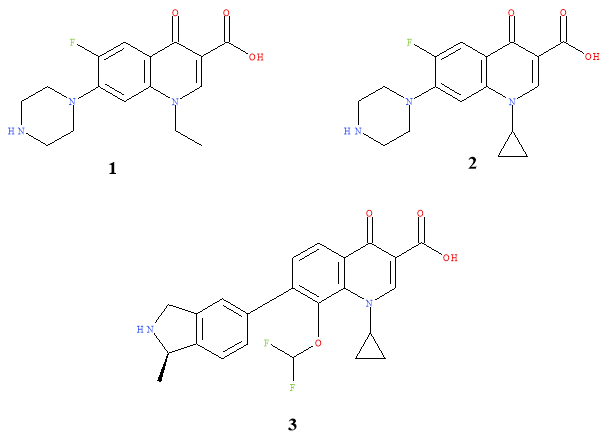
Figure 4:Generations of quinolone drugs.
Three modified quinolone cores have acted as templates for drugs that are on the commercial market. The cores were 4-oxo-1,4-dihydroquinolone (4), 7-oxo-2,3-dihydro-7H-pyrido-[1,2,3-d,e]-1,4-benzoxazine (5) and 4-oxo-1,4-dihydro-[1,8]-naphthyridine (6). These selected cores are illustrated in Figure 5.3
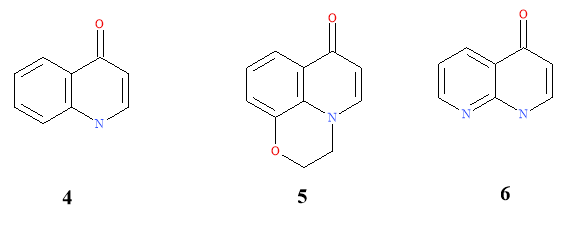
Figure 5: Cores used as templates for commercial drugs
Retrosynthesis:
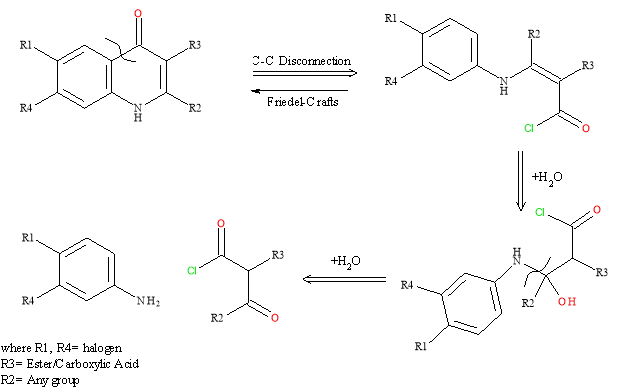
Scheme 1: Retrosynthesis of 4-Quinolone core6
A carbon-carbon disconnection between the ketone and the aromatic ring, the reverse would be a Friedel-Crafts reaction. The double bond is opened and the hydroxyl group that is added is converted to a carbonyl group. The final disconnection, N-C, results in the starting materials; a keto ester and the substituted aniline.
Synthesis:
Several synthetic approaches have been taken to generate the 4-quinolone core. The Gerster-Hayakwa and Chu-Mitscher reactions are used specifically for the synthesis of the drug Levofloxacin. The Chu-Li route was established primarily for 9-cyclopropylpyrimidinones. The Gould-Jacobs reaction, Grohe-Heitzer cycloacylation and Conrad-Limpach-Knorr are appropriate methods of synthesising the generic 4-quinolone core.3
The Conrad-Limpach-Knorr synthesis will generate quinolones but this reaction will give different products depending of the whether it is kinetically or thermodynamically controlled. Aniline and 3-keto ester are mixed and at room temp the keto group joins the nitrogen of the aniline giving an enamino ester (kinetic product) and cyclisation at 250°C results in a 4-quinolone. Thermodynamically controlling this reaction, by carrying it out at 140°C, results in an amido-ketone dominating regardless of the less reactive ester on the keto ester being the centre of the first nucleophile attack. Ring closure results in a 2-quinolone.7
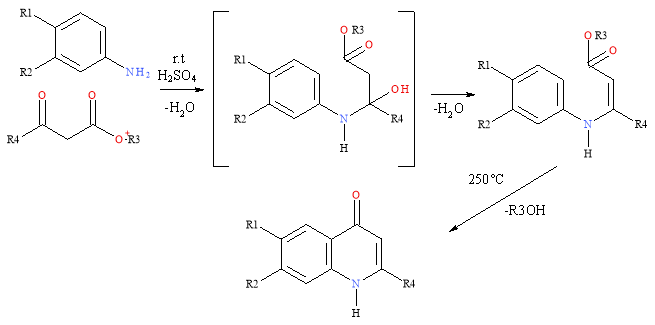
Scheme 2: Conrad-Limpach-Knorr Synthesis (kinetically controlled)
As the substrate for the cyclisation needs to be the high-energy tautomer and cyclisation causes loss of aromaticity in the ring, solvents with high boiling points are generally used in this synthesis.8 The reactions are encouraged by electron-donating substituents in the aromatic ring including methoxy or amino groups. These give increased yields in the condensation and ring closure steps. A CF3 group can act as an acceptor at C-4.9 The short reaction sequence limits possibility of loss of yield.
Rational Drug Design:
Illustrated in Scheme 3 is the process of rational drug design. Computational screening is used to identify the target and generate the lead compound. This is modified considering the biological aspects, the 3D structure, the QSAR and reactivity of the compound. This generates a new lead which is optimised and put forward for preclinical trial.
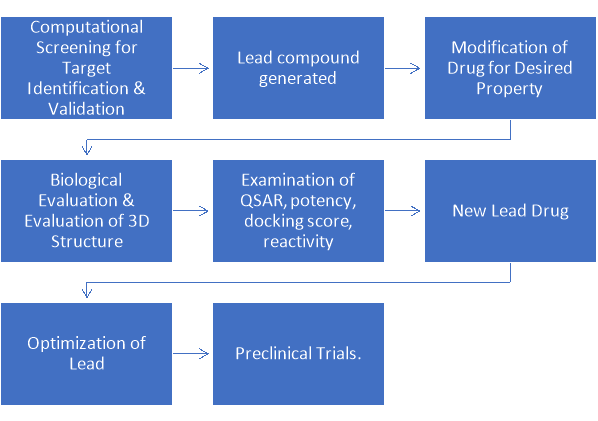
Scheme 3: Rational Drug Design Process10
Nilsen et al. used endochin as a lead for optimisation in a rational drug design study. The target selected was the multiple stages of the life cycle of malaria. Endochin is potent against malaria but is not active in vivo due to rapid and extensive metabolisation. Optimisation was required to form endochin-like quinolones (ELQ) that retained the activity of endochin but were biologically active. The aims for optimisation was to improve metabolic stability and aqueous solubility as well as eradicate cross-resistance.11
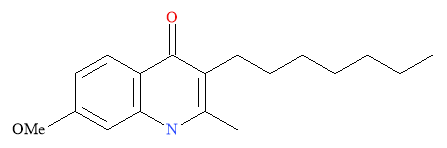
Figure 6: Structural Representation of Endochin
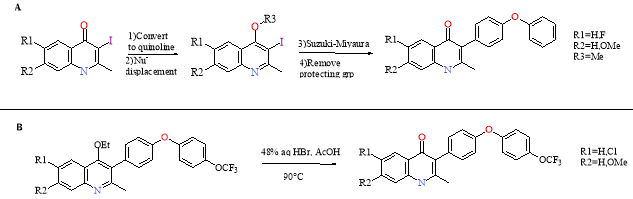
The EQLs were synthesised by converting the quinolones to quinolines, followed by nucleophilic displacement. The quinoline undergoes Suzuki-Miyaura coupling with a boronic ester and finally the protecting group is removed (Scheme 4 A). An OCF3 group was also added to the side chain for further optimisation (Scheme 4 B).
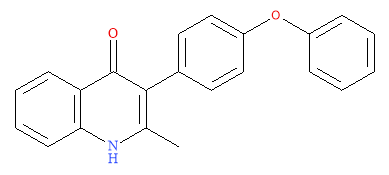
An orally active class of quinolones were synthesised; 4(1H)-quinolone-3-diarylethers. The initial lead, ELQ-233 (Figure 7: ELQ-233Figure 7), displayed low nano-molar IC50 values. The optimisation step was to introduce an aryl group at C-3. A lipophilic diphenylether side chain was used as it had been previously reported as integral in other antimalarial drugs. This was to work with the methyl group at C-2 to cause out of plane movement of the sterically large aromatic ring, altering the π−π stacking from the numerous H-bonds. This variation would not be perused as ELQ-233 was equipotent to endochin.
Due to the success of adding a fluoro group to the quinolone, to further build on the optimisation of endochin, a fluorine was added at C-6 on a second optimised molecule (7) along with a methoxy group at C-7 (as is in the endochin structure); illustrated in Figure 8.11 The methoxy group is a useful substituent due to its lipophilic and hydrophilic components in close proximity.
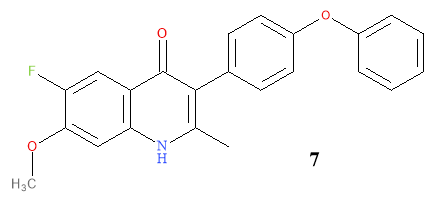
Figure 8: Illustration of Compound 7
However, both ELQ-233 and 7 were metabolically unstable and therefore did not fulfil the optimisation requirements.
Table 1: Values obtained for the optimised molecules
|
Compound |
cLogP |
EC50 (nm) |
|
Endochin |
3.35 |
3.8 |
|
ELQ-233 |
3.70 |
8.4 |
|
7 |
3.73 |
40 |
|
8 |
5.66 |
2.2 |
Further derivatives were generated and the pattern that emerged indicated that the substitution pattern on the aromatic ring influenced the reactivity with malaria. This result led to the rational design of further ELQ derivatives. Straight-forward reactions were continued to be used. The boronic ester with a varying diarylether side chain undergoes palladium mediated coupling with the quinolone then a demethylation occurs using hydrobromic acid to give the desired product. Repositioning the OCF3 group to the side chain increased the efficacy and the metabolic stability. The compounds that was found to be metabolically stable and potent had a chloro-group at C-6 and OCH3at C-7.
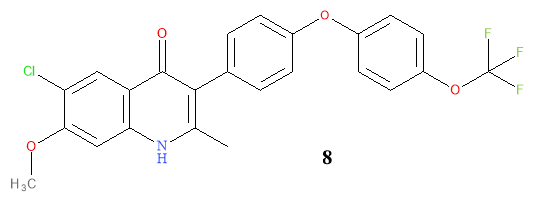
Figure 9: Structural representation of compound 8
Although compound 8 was the most potent compound, the high logP value is a disadvantage as it does not follow Lipinski’s Rule of Five which is the basis for most developed drugs. To improve solubility and allow for lower dosages of 8, bioisoteres of the side chain were employed. The OCF3 was replaced by CF, Cl and F by Nielson et al. and displayed subnanomolar activity.
Other options would be to double the terminal OCF3 group, double the substituents on the diaryl side chain, convert the diphenyl ether side chain to a dipyridine ether side chain or replace a phenyl ring in the side chain with a cyclopropane group. Phenyl rings can be replaced by a heteroaromatic ring or a saturated ring to improve efficacy, lipophilicity and specificity of binding. The introduction of a pyridine ring should reduce the metabolism of the phenyl ring and toxicity of metabolites.12 In heterocycles, metabolism can be more complicated with hetero-atoms being oxidized and/or ring opening reactions possible- slowing metabolism. Cyclopropane was explored as derivative of the phenyl ring resulting in compounds with reduced molecular weights and lower lipophilicities. It also limits the conformations available and increase yields of ELQs.11Scheme 5 below are the same coupling reactions that are stated by Nielson et al. but with the suggested changes to further improve the lead. Caution must be taken when adding substituents to the side chain so that Lipinski’s rule of five is obeyed; there must be no more than 5 hydrogen bond donors or 10 hydrogen bond acceptors and the molecule should be below 500 Da.
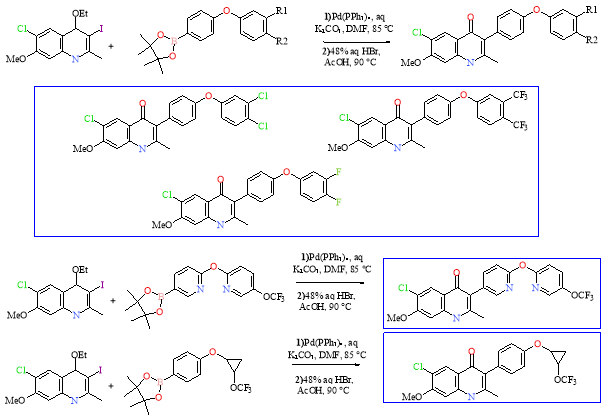
Scheme 5: Suggested further optimised lead molecules
Rational drug design is an advantageous modification method as it is a streamline process when compared with SAR or QSAR as there is no trial and error, all leads and derivatives are prepared having been predicted by computational means previously. The computational aspect allows for all compounds and potential targets to be envisaged in 3D before they are synthesized. This computed information is then stored on large databases which can assist future drug development work. Rational drug design can be an expensive technique as a specialized team is required with knowledge in biology, chemistry and computer science. Costs rise due to payment of wages for the team and the specialized equipment and computer software that is vital. Although the computational aspect of this method is beneficial it can also be a disadvantage as not all predicted compounds can be synthesized and if the compounds are synthesized they may not act as predicted when in vivo. Specifically, in this rational drug design study, the reactions utilised readily available reagents that were also inexpensive. They were straight-forward reactions that gave high yields and could be scaled up. These are vital as the cost of antimalarial drugs must be kept down so that all people can afford to access it.11
Structure-Activity Relationship (SAR):
SAR studies examine how the structure of the molecule effects the activity. SAR considers structural characteristics and relates them the activity therefore it is necessary to have a well characterized database to compared the results against. The basic principle of SAR, that structure determines properties and reactivities in a biological system, is of importance when determining toxicological properties. This is of huge significance for quinolone development as they must be nontoxic in vivo while remaining bacteriosidal.13
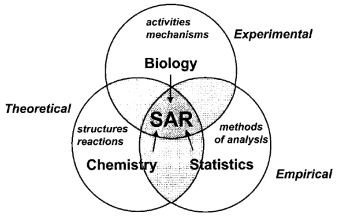
Figure 10: Areas SAR Studies consider13
These studies examine which modifications are possible to the core ( Figure 11) and which substituents cannot be modified without negatively interfering with the activity and potency of the drug. There can be qualitative and quantitative aspects to these studies. The quantitative considerations are part of a quantitative structure activity relationship (QSAR) which will be discussed later.
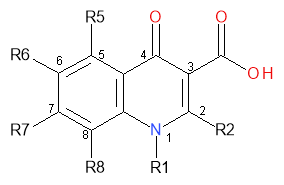
Figure 11: Quinolone core positions for the SAR study
Table 2: Important Positons on the Quinolone core-SAR Study results14
|
Position |
Influence On: |
Preferred substituent: |
Effect of substituent: |
|
1 |
The pharmacokinetics and has control on overall potency. |
Cyclopropyl |
increase activity against gram negative microbes |
|
5 |
Activity against gram-positive bacteria. |
NH2 and CH3 moiety |
improve activity against gram-positive bacteria |
|
7 |
Spectrum of activity and pharmacokinetics. |
5/6 membered N heterocycle (aminopyrrolidines and piperazines) Alkyl group |
Aminopyrrolidines: increase activity against gram-positive bacteria Piperazines: increase activity against gram-negative bacteria. Alkyl: enhance gram- positive potency and lengthen the serum half-life |
|
8 |
Pharmacokinetics and specific activity on anaerobes. |
CF, CCl and COMe |
improve activity against anaerobic bacteria. alter specific interaction of the agent in vivo. |
SAR studies reveal that a hydrogen at R2 is preferred. Any larger moiety would likely cause steric hindrance with the adjacent carboxyl group at C-3 and the oxygen at C-4. These substituents are vital for activity as these positions are where binding to DNA bases occur before the sites are made available for other hydrogen bonding to proceed by DNA gyrase. A small molecule is best for potency at R6. This is usually a fluorine group in later generations of quinolones as it produces molecules of between 5 to 100 times greater potency that when a hydrogen is positioned at R6.14
Ciprofloxacin (Figure 12) is one of the original patented quinolone anti-bacterial agents. It is first generation as it has moderate activity towards gram negative bacteria, poor pharmacokinetics and poor bioavailability.
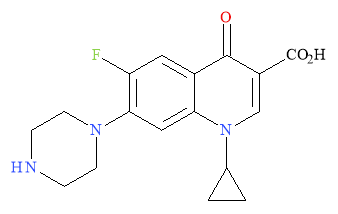
Figure 12: Structural representation of Ciprofloxacin
However, using the results of the SAR, Ciprofloxacin can be further optimized. Scheme 6 utilizes straight-forward reactions to attach an -NH2 moiety to the core at positon 5 where the SAR study indicates it has the greatest influence. Firstly, nitration of the aromatic ring occurs, followed by a reduction of the nitro group to an aniline with palladium on carbon.
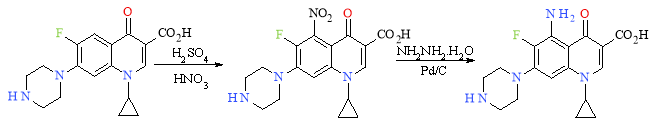
Scheme 6: Further optimisation based on SAR Study Results
The new lead is still in agreement with the SAR results as the carboxyl and oxygen are present at C-3 and C-4 respectively. The preferred substituent as stated in Table 2 are also used throughout the reaction scheme; the piperazine is at position 7, the fluoro group at positon 6 and the cyclopropyl group at positon 1. These substituents can positively influence the spectrum of activity the potency and the overall pharmacokinetics of the molecule. The addition of extra hydrogen bond acceptors/donors must be limited so as not to disobey Lipinski’s rule of five by having more than 5 hydrogen bond donors or 10 hydrogen bond acceptors.
These reactions, like the rational drug design reactions, utilize readily available, generally inexpensive reagents which is important to keep the cost of the anti-malarial drug down. Palladium is an exception as it is a rare metal but cheaper alternatives could be used for this step such as Raney Nickel although this generates intermediates before the aniline is formed unlike the direct formation when the palladium catalyst is used.15
SAR studies can represent molecules as 2D, atoms and bonds, or 3D, steric effects and electrostatics. 3D is best for when the receipt-mediated mechanism is known. Successful SAR studies also need appropriate methods of analysis which depend on whether quantitative or qualitative analysis is being perused and if the mechanism is known. The ideal SAR model should have adequate molecules for fair statistical analysis, a wide range of activities and an even distribution of molecules in each compound class. This model is rarely found when toxicology is considered.13
Quantitative Structure-Activity Relationships (QSAR):
The basic principle of QSAR is that similar molecules have adequate similar mechanistic elements so that a common rate-determining step is shared among them and that they have comparable energy requirements for activity. This principle is taken further and the assumption is that differences in reaction rates results in differences in activity or potency.13 Lipophilic, electronic and steric effects are considered in QSAR studies. QSAR provides an equation that quantifies the SAR and allows for predictions about which property has an important role in the distribution/ mechanism of the drug. Predictions cut down on the volume of analogues to be synthesised. Equations are only applicable to compounds of the same structural class. Outliers indicate when a feature is important and can produce new leads. The QSAR may not give accurate predictions as the parameters have covariance on each other; the predicted model may vary in vivo.6
Lipophilicity/Hydrophobicity:
This can be considered as the lipophilicity of the molecule or the lipophilicity of the substituents attached. Partition Coefficient, P, is the parameter associated with the lipophilicity of the molecule and is measured by Equation 1.

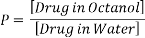
Equation 1: Representation of the lipophilicity parameter. Solvents chosen to represent the Central nervous system
The activity of a molecule can be related to the P value as a molecule must be able to cross membranes and be transported through the body to its target site which is dependent on its lipophilicity. Varying substituents on the core can alter the P value in whichever direction is more beneficial for the activity of the molecule. From SAR studies increased quinolone activity occurs when a lipophilic substituent, such as a halogen, is attached at C-6. Simple reactions, including nitration and chlorination (Scheme 7), will add these substituents to the core, again using cheap and readily available reagents. In general, increasing lead hydrophobicity increases activity (Figure 13). This does not go to infinity as there is a point at which the lead is to hydrophobic to be transported in vivo.6 LogP should not be more than 5 per Lipinski’s rule of 5; high enough to bind, low enough to be released.
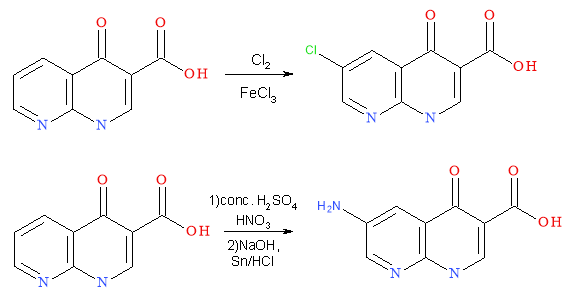
Scheme 7: Addition of lipophilic substituents to the quinolone core to alter the P value
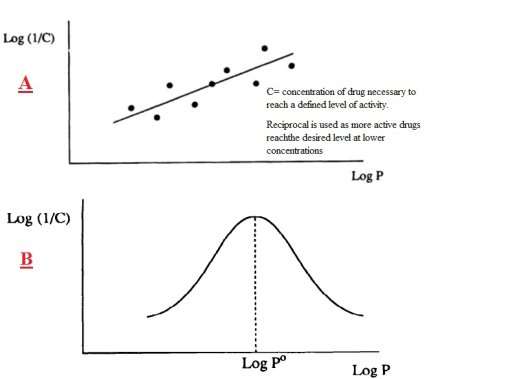
Figure 13: Linear relationship between biological activity 1/C and lipophilicity6
Electronics:
This parameter can influence ionisation and polarity which alters how a drug passes through a membrane or how strongly it binds to a receptor. Hammett substitution constant (σ) is the measure of the electron withdrawing or donating ability for substituents on an aromatic ring. σ affects the equilibrium and the value is dependent on induction/resonance effects and whether the substituent is para or meta directing. Ortho is not considered due to sterics.6
Table 3:Substituents that can alter the σ parameter
|
Electron Donating Group (para): |
Electron Withdrawing Group (meta): |
|
-NH2 |
-NO2 |
|
-OH |
-CONH2 |
|
Halogen |
-CN |
Generally, electron-withdrawing substituent, positive σ values, increase activity (Table 3). Simple reactions like those illustrated in Scheme 7 are used to attach the σ-influencing substituents to the quinolone.
Sterics:
Drug molecules must approach and successfully bind to a receptor and the sterics of the molecule can alter this approach. Bulk can result in nonbinding as the drug is sterically hindered from approaching the target site. It can also limit the available conformations so that only the most efficient arrangement binds to the receptor.
Table 4: Parameters for measurements of steric effects6
|
Measure of Steric Effect: |
Key Feature: |
Other Factors: |
|
Taft’s Steric Factor (Es) |
Quantifies steric feature of substituents |
Limited to use on certain substituents |
|
Molar Refractivity (MR) |
Measures volume occupied by atom(s) |
Corrects for ease of polarisation. |
|
Verloop Steric Parameter |
Computer programme calculates steric values |
For use with any substituent |
Using the quinolone core, modifications can be made so that the sterics prevent rapid metabolisation of a drug molecule in vivo which will extend its half-life and lead to better activity. Binding the quinolone to a large side chain restricts it from binding to smaller sites which Nilsen et al. conclude lead to better selectivity.11Scheme 8 illustrates the addition of bulky side chains that can give better selectivity as they will only approach the sites they fit into. Furthermore, the double bond linkage and aromatic rings restrict the conformation that the molecule can adopt, increasing selectivity. Bulky side chains can prevent rapid metabolism occurring. When adding bulk, caution must be taken to ensure the molecule stays below the recommended 500 Da.
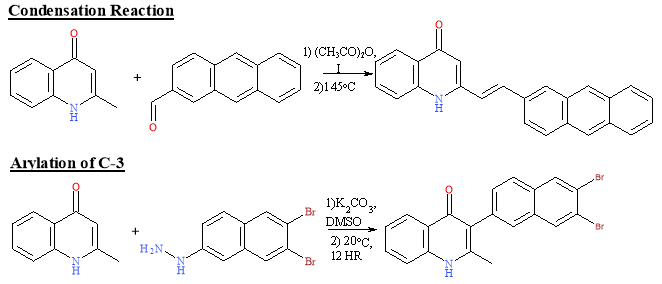
Scheme 8: Reactions to alter the sterics of a quinolone core16
3D-QSAR:
3D-QSAR considers the relative spatial arrangement of model compounds and aims to correlate the features across molecules that affect activity and are required for ligand binding. 3D-QSAR studies the geometry, pharmacophore and molecular field. Key assumptions of 3D-QSAR13:
- The model compound and not its metabolite cause the biological response.
- The studied conformation is bioactive.
- Solvent effects are not considered
- The system is in equilibrium
- All compounds bind in the same manner to the one target.
3D-QSAR puts compounds with common configurations in a 3D grid, calculates the interaction and tabulates the results. An equation is then created based on the relationship between the calculations and the reported values. This verifies QSAR results. Conformers are superimposed to display the ‘common ligand-binding orientation to the receptor.’ ‘Probe atoms’ calculate steric and electrostatic fields.13 3D-QSAR studies on 1,3,5-triazine, quinolone derivatives, determined less bulky groups on the heteroatom ring, more bulk on the aromatic ring and a less electronegative substituent at position 6 results in improved antibacterial activity.17 This method is not yet well developed enough to be a reliable standalone source of modifications.
Quinolones are a well-developed class of antimicrobial agents. Currently there are four generations of quinolones- classified based on their activities in vivo. Fluoroquinolones are the latest developments in the quinolone class. They have improved activity against gram negative bacteria and are potent against gram positive microbials that earlier generations were not. The retrosynthesis of the quinolone core is simplistic with common starting materials; a ketoester and aniline. Several synthetic approaches will generate the 4-quinolone core. The Conrad-Limpach-Knorr synthesis was chosen to examine in detail as it only requires readily available reagents and heat and has minimal steps minimising the opportunities for loss of product to occur. A disadvantage to this reaction is the mix of products that is possible. Kinetic control results in a 4-quinolone core, thermodynamic control will generate 2-quinolone. Modifications to the quinolone core can be made via rational drug design, SAR studies, QSAR studies and 3D-QSAR. 3D-QSAR is not as developed as the other methods. All reactions and suggested variations to the core, by the other methods, are all straightforward reactions with inexpensive reagents. It is important to keep the cost of antimicrobial drugs down so that everyone can have access to them. SAR, QSAR and rational design all provide evidence for the inclusion of a fluoro group at C-6 which agrees with the current interest in fluoroquinolones. Addition of bulk to limit conformations and prevent metabolism accompanied by halogen substituents improve activity by altering the hydrophobicity/lipophilicity, steric and electronic effect. Rational drug design is a strong choice for the modification of quinolones as it is a streamlined, computer based process, which saves on time and money. Furthermore, it uses information we already have as a foundation so all not all modifications need to be novel. Simple, robust reactions can result in optimisation.
References
1 S. Emami, A. Shafiee and A. Foroumadi, IJPR, 2010, 4, 123-136.
2 K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry (N. Y.), 2014, 53, 1565-1574.
3 C. Limberakis, in The Art of Drug Synthesis, ed. D. S. Johnson and J. J. Li, John Wiley & Sons, Inc., New Jersey, 2006, p. 39-69.
4 A. M. Emmerson and A. M. Jones, J Antimicrob Chemother, 2003, 51 Suppl.1, 13-20.
5 P. Ball, J. Antimicrob. Chemother., 2000, 46, 17-24.
6 G. L. Patrick, An introduction to medicinal chemistry, Oxford University Press, 2001.
7 M. Sainsbury, in Heterocyclic Chemistry, ed. E. W. Abel, The Royal Society of Chemistry, 2001, p. 48-49.
8 J. Brouet, S. Gu, N. P. Peet and J. D. Williams, Synthetic Communications, 2009, 39, 1563-1569).
9 G. Strohmeier, W. Fabian and G. Uray, HCA, 2004, 87, 215-226.
10 S. Mandal, M. Moudgil and S. K. Mandal, Eur. J. Pharmacol., 2009, 625, 90-100.
11 A. Nilsen, G. P. Miley, I. P. Forquer, M. W. Mather, K. Katneni, Y. Li, S. Pou, A. M. Pershing, A. M. Stickles, E. Ryan, J. X. Kelly, J. S. Doggett, K. L. White, D. J. Hinrichs, R. W. Winter, S. A. Charman, L. N. Zakharov, I. Bathurst, J. N. Burrows, A. B. Vaidya and M. K. Riscoe, J. Med. Chem., 2014, 57, 3818-3834.
12 N. A. Meanwell, J. Med. Chem., 2011, 54, 2529-2591.
13 J. D. McKinney, A. Richard, C. Waller, M. C. Newman and F. Gerberick, Toxicol Sci., 2000, 56, 8-17.
14 G. S. Tillotson, J. Med. Microbiol., 1996, 44, 320-324.
15 J. Wisniak and M. Klein, Ind. Eng. Chem. Prod. Res. Dev, 1984, 23, 44-50.
16 M. Ravi, P. Chauhan, R. Kant, S. K. Shukla and P. P. Yadav, J. Org. Chem, 2015, 80, 5369-5376.
17 R. L. Sawant, S. S. Ramdin and S. R. Rathi, Int J PharmTech Res, 2012, 4, 1096-1100.