Multifunctional Porous Organics Synthesis
|
TITLE OF WORK |
The synthesis and characterisation of functionalized porous organics |
Abstract
Lophine based radicals have been studied for many years due to their photochromic nature, recent literature has seen attempts to connect two radicals to carbon based backbones. In this report we will discuss attaching six lophine molecules to a single backbone which is a phosphazene ring. Cyclophosphazene rings have a unique conformation allowing for supramolecular architectures to be formed by varying the substituents, these architectures range from 0-2D structures. The hexalophine molecule exhibits 1D channels due it’s unique conformation which leads to the formation of a porous material. Porous organics are of high interest in recent literature due to them being a cheaper, less toxic alternative to MOF’s as well as having the ability to be modified and tailored easily due to the simple starting materials. In this report we shall state synthetic pathways from simple molecular building blocks to hexalophine (32%) and two derivatives, compound 4 (46%) and 5 (15%). These two derivatives have the potential to increase pore stability. This report will also include the oxidation of hexalophine to its radical species which were found to display photochromic properties and it is also believed that this material could contain ‘light gated’ pores which are closed in the dimer form of the product but then open upon irradiation of UV of 365nm.
1. INTRODUCTION
1.0 Properties and potential uses of Lophine
This report will feature chemistry based upon the triphenylimidazolyl based radicals (TPIRs) as well as the dimer form hexaarylbiimidazole (HABI). Lophine (2,4,5-triphenyl-1H-imidazole) is a heterocyclic imidazole derivative where three phenyl rings are attached to the imidazole ring (figure 1), these imidazole derivatives are an area of importance due to the many interesting properties it possesses. Lophine has been of interest to researchers for many years with its chemiluminescent properties being first discovered in 1877 by Radziszewski.1 The oxidation of the lophine molecule follows the reaction scheme below, where lophine is oxidised to the TPIR radical state via addion of base and treatment with Fe forming one of six dimers both in solution and the solid state (figure 2). These materials display photochromic, thermochromic and piezochromic properties. The mechanism of photochromism of the lophine dimer is the homolytic cleavage of the 2C-1’N bond of the dimer by irradiation with light, forming two lophine radicals.2 The fast colour change associated with this dimer opening means there is a potential use in display screens or as the basis for molecular switches, where irradiation with light opens the dimer forming the radical in turn switching the external component On/Off. These possible applications have caused a focus on creating methods to manipulate and control the colour of these materials and as well as to increase the rate of switching.3 The colour of the radical form is dependent upon the substituents on the ring, the more conjugated the system the further toward the red side of the spectrum the colour.4 Lophine displays piezochromic properties in its solid state, upon applying pressure lophine crystals the colour change of yellow to blue/green has been observed, this occurs via the radical dissociation of the 1N-1’N dimer bond.5 Other more unusual modes have been of dimerization such as the 2C-2’C have now also been isolated, this mode features a unusually long C-C bond.3
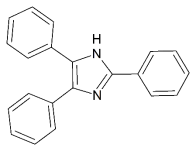
Figure 1 – The structure of Lophine (2,4,5-triphenyl-1H-imidazole)
Another interesting lophyl radical derivative is the 1,8-TPID-naphthalene radical which consists of 2 lophine radicals connected by a naphthalene ring. 1,8-TPID-naphthalene has been characterised in the solid state by Abe et al; who studied the photoreactivity of the compound. 1,8-TPID-naphthalene allowed the group to fix two radicals onto a backbone meaning that the radicals were in close proximity and could dimerise easily as opposed to two radicals not connected via a backbone which first need to ‘find eachother’ in order for dimerization to occur (figure 3). They found that the dimer could be cleaved photochemically giving the diradical and then could be thermally converted back to the dimer.6 They also found that the formation of a peroxide bridge was much more common than previous literature had accounted for. This bridging occurred when the 1,8-TPID-naphthalene radical was placed under an O2 atmosphere.7 This peroxide bridge formation is very important as when oxygen quenches the spin to form the closed shell peroxide it does so irreversibly meaning the material is no longer photochromic. It was then shown that this peroxide degradation pathway can be generalised for the chromic dimers of TPIR materials, with non-tethered TPIRs forming similar peroxide bridges.8 A recent development by the same research group has also shown how a thiophene substituted phenoxyl-imidazole radical complex (PIC) can generate two non-equivalent radical upon irradiation with UV light. The colour change can be fine-tuned from tens of seconds to nanoseconds, however a sensitivity to lower wavelengths of light meant that applications were limited. This problem was rectified 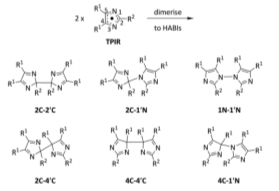 by adding a phenyl group to the 5 position of the thiophene ring.9
by adding a phenyl group to the 5 position of the thiophene ring.9
Figure 2 – The six potential dimerization modes of two lophyl radicals.3
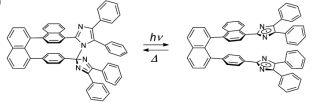
Figure 3 – Illustrates the reversible conversion of 1,8-TPID-naphthalene between its radical and dimer forms.
1.1 Hexa-substituted Phophazene Rings, Tectons and Crystal Engineering
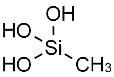
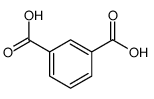
A key component of this project was the exploitation of the Phosphazene ring as ‘soft’ tecton; a tecton is a molecular building block that interacts with ‘sticky’ sites, formally known as supramolecular synthons, via spatial arrangements to induce the formation of supramolecular aggregates. The search for new tectons is a hot area of research due to the constantly growing field of crystal engineering where the aim is to produce functional single crystal materials using intermolecular interactions. The arrangement of molecules in solid state structures is largely dependent on striking a fine balance between intramolecular forces and packing interactions, knowledge of these forces is key to the field of crystal engineering.10 In general, ‘hard’ tectons interact via more robust synthons than ‘soft’ tectons and therefore usually crystallise with unambiguous geometries. Whereas ‘soft’ tectons and less robust synthons allow for the formation of supramolecular isomers (figure 4).11 These tectons allow for many functional materials to be formed in a one pot synthesis, they also have the ability to be carefully adjusted to alter and improve upon their functionality.12 Cyclophosphazenes are extremely ‘soft’ tectons that interact with ‘soft’ synthons to give a large variety of supramolecular architectures in the solid state. The conformation of the cyclophosphazenes is also very interesting as three substituents reside above the plane of the ring and three below (figure 5). It was found that small modifications to the substituents attached to the ring gave supramolecular architectures ranging from 0D to 2D structures (figure 6) such as include monomer, dimer, cyclic hexamer, zigzag chain, linear chain, double chain, graphite-type sheet, rectangular grid and hexagonal close-packed sheet. Such variety of structures came from the easy rotation about the exocyclic P-N bonds, which allowed variable directionalities for all of the N-H bonds. 11
3a) 3b)
Figure 4a) Shows benzene-1,4-dicarboxylic acid a classic example of a hard tecton. 4b) shows a silanetriol a classic soft tecton.
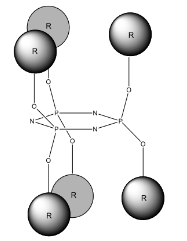
Figure 5 – Conformation of a Hexa-substituted Phosphazene Ring.
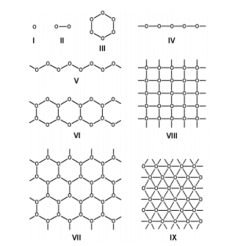
Figure 6 – Schematic representations of aggregation patterns of (RNH)6P3N3 in the solid state.11
1.2 Magnetism and Magnetic Frustration
The materials we aim to make during this project once appropriately oxidised should go on to form dimers which when irradiated with UV light form discrete radicals containing unpaired electrons thus giving each radical an associated magnetic moment. Neutral organic radicals tend to be paramagnetic and exhibit Curie- Weiss behaviour, where the spin vectors of the unpaired electrons are randomly aligned above the Curie temperature (Tc), which is the critical temperature below which the long ordered state is established, unless a magnetic field is applied. If spin vectors are parallel to each other below the Curie temperature then the material is ferromagnetic, if the spin vectors are aligned antiparallel to one another the material is antiferromagnetic.13
Due to the geometry of the phosphazene ring (three substituents pointing up above the plane of the ring and three below) there is the possibility of forming a 2D triangular lattice of spins if the substituents are organic radicals. This could possibly lead to geometric magnetic frustration due to the fact that the two nearest neighbours to a spin are themselves nearest neighbours and therefore antiferromagnetic couplings cannot be satisfied (figure 7).14 This can lead to very interesting magnetic properties. Most solid state examples of this geometric frustration tend to be transition metal oxides, however, studies have been done on organic systems that also distribute geometric frustration.13 One example of this is m-MPYNNP+ which is a spin ½ organic radical that upon dimerization forms a 2D triangular lattice which demonstrates the aforementioned geometric frustration.14
The issue of spin frustration has been around for a long time as Quantum spin liquids (QSL) were first theoretically proposed by Anderson many years ago which are now a hot topic of research due to the realisation of these QSL’s in organic materials such as k-(ET)2-CH2(CN)3, which has a near perfect triangular lattice with angles very close to 120°.15 A QSL is an exotic ground state where interacting spins continuously fluctuate with no formation of low range magnetic order even at sufficiently low temperatures.16


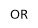



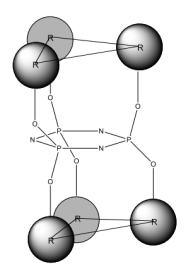
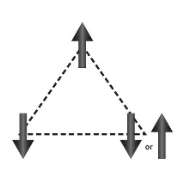




Figure 7 – Antiferromagnetic coupling cannot be fulfilled causing geometric magnetic frustration.
1.3 Porous Materials
Porous materials are as important as ever due to their wide spread use in several fields such as catalysis and gas absorption,17 meaning they can play a critical role in the route to solving our growing energy shortage problems. Porous materials are instantly associated with materials such as zeolites, metal organic framework (MOF) and organic polymers. MOF’s are characterised by their tuneable pores and inherent flexibility which more classical carbon or oxide based structures do not possess, this gives a wide range of applications for MOF’s such as gas storage, separation, drug delivery or catalysis.18 Porous organics have been discovered in nature but are largely created synthetically and approaches often need to be coupled with knowledge of crystal engineering. These materials are often held together by directional forces which form extended frameworks of noncovalent interactions such as π- π interactions, hydrogen bonding or coordinate bonding (figure 8).17 Like zeolites and MOFs porous organics have the ability to selectively absorb atoms and molecules in the gas phase.19 There are many positives to these porous organics, they are often cheaper to synthesise and less toxic as well as being far less dense than metal containing MOFs.
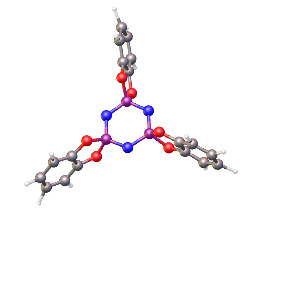
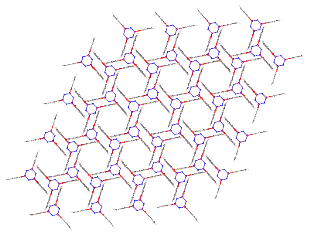
Figure 8 – One example of a Porous Organic by Sozzani et al.
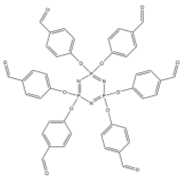
1.4 P3N3 Hexalophine
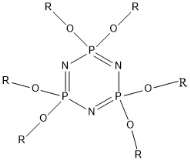 The chemistry of P3N3 Hexalophine incorporates much of the chemistry discussed above and has many interesting properties. The P3N3 Hexalophine molecule consists of six lophine substituents attached to the phosphazene ring via a P-O bond (figure 9). Previous work in the Robertson group found that the solid state structure of Hexalophine consisted of a pseudo hexagonal motif giving rise to 1D channels that run parallel to the stacking axis (figure 10). The molecule maintained its D3 symmetry in the solid state. The phenyl rings twist to maximise pi-stacking interaction and the 3 imidazole nitrogen atoms form hydrogen bonds to a central water molecule. There are 4 pi-pi interactions that stabilise this structure, the molecules are in a ‘slipped’ conformation allowing the phenyl and imidazole to rings overlap. Other non-covalent interactions such as van der Waals interactions also help to stabilise the structure. The high symmetry of the structure which comes from the hexa substituted phosphazene ring (three above the plane of the ring and three below) gives an equal distribution of intermolecular forces giving a high probability of forming a stable porous structure. IGA measurements with CO2 loading at 195K shows a Type I isotherm with absorption reaching 8cm3g-1 at a pressure of 700mmHg confirming the materials’ microporosity. However only a small fraction of the void space contained CO2.
The chemistry of P3N3 Hexalophine incorporates much of the chemistry discussed above and has many interesting properties. The P3N3 Hexalophine molecule consists of six lophine substituents attached to the phosphazene ring via a P-O bond (figure 9). Previous work in the Robertson group found that the solid state structure of Hexalophine consisted of a pseudo hexagonal motif giving rise to 1D channels that run parallel to the stacking axis (figure 10). The molecule maintained its D3 symmetry in the solid state. The phenyl rings twist to maximise pi-stacking interaction and the 3 imidazole nitrogen atoms form hydrogen bonds to a central water molecule. There are 4 pi-pi interactions that stabilise this structure, the molecules are in a ‘slipped’ conformation allowing the phenyl and imidazole to rings overlap. Other non-covalent interactions such as van der Waals interactions also help to stabilise the structure. The high symmetry of the structure which comes from the hexa substituted phosphazene ring (three above the plane of the ring and three below) gives an equal distribution of intermolecular forces giving a high probability of forming a stable porous structure. IGA measurements with CO2 loading at 195K shows a Type I isotherm with absorption reaching 8cm3g-1 at a pressure of 700mmHg confirming the materials’ microporosity. However only a small fraction of the void space contained CO2.
Figure 9 – The structure of Hexalophine.

Figure 10 – The crystal structure of Hexalophine, illustrating the 1D channels.
1.5 Aim of Project
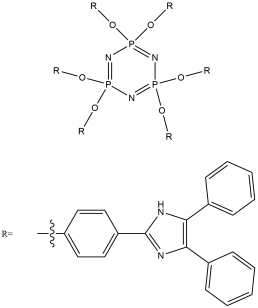
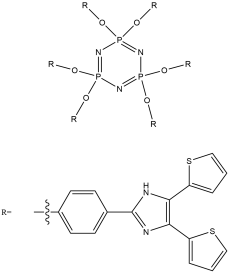
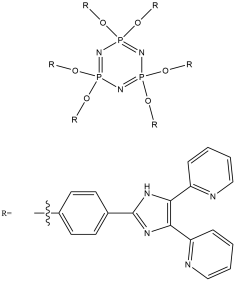
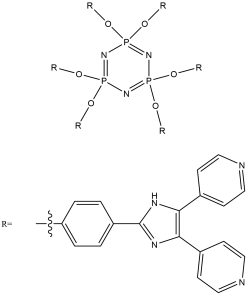
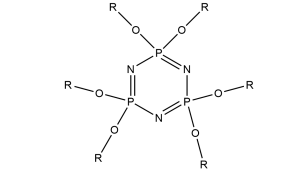 The aim of this project is to combine our knowledge of the chemistry of both lophine radical systems and cyclophosphazene rings to find elegant synthetic routes which will improve upon the porous properties of hexalophine. This will be done by varying the ring substituents to increase stability of the pores and to improve its gas absorption properties. Three lophine derivatives have been identified as plausible candidates for thid are where two of the phenyl groups on the lophine will be replaced by two thiophene rings and two pyridine rings respectively, with the position of the nitrogen in the pyridine ring being varied depending upon the synthetic pathway (figure 11). We wish to synthesise and fully characterise these derivatives including growing single crystals for structural studies via X-ray crystallography. Also a stoichiometric controlled oxidation of the hexalophine material to its neutral radical/dimer pair is required in order to further investigate the properties of hexalophine such as the possibility of creating a photochromic material that has ‘light gated’ pores that open on irradiation with UV light.
The aim of this project is to combine our knowledge of the chemistry of both lophine radical systems and cyclophosphazene rings to find elegant synthetic routes which will improve upon the porous properties of hexalophine. This will be done by varying the ring substituents to increase stability of the pores and to improve its gas absorption properties. Three lophine derivatives have been identified as plausible candidates for thid are where two of the phenyl groups on the lophine will be replaced by two thiophene rings and two pyridine rings respectively, with the position of the nitrogen in the pyridine ring being varied depending upon the synthetic pathway (figure 11). We wish to synthesise and fully characterise these derivatives including growing single crystals for structural studies via X-ray crystallography. Also a stoichiometric controlled oxidation of the hexalophine material to its neutral radical/dimer pair is required in order to further investigate the properties of hexalophine such as the possibility of creating a photochromic material that has ‘light gated’ pores that open on irradiation with UV light.
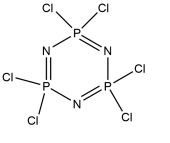

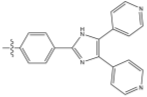
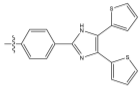
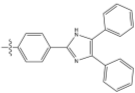
R= or or
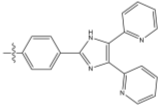
Figure 11 – the synthetic targets of this project.
2. Results and discussion
2.0 Hexa-benzaldehyde Synthesis
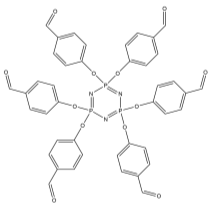
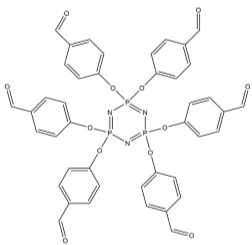
The first step in this synthetic project involved attaching six benzaldehyde units to the phosphazene ring via a P-O oxygen bond. This was achieved by reacting the phosphonitrillic chloride trimer along with 6 equivalents of 4-hydroxybenzaldehyde, potassium carbonate in dry THF (figure 12). The reaction was monitored with 31P NMR which showed completion when only a singlet was present in the spectra indicating full substitution. This produced the compound 1 which was the precursor for many of the subsequent reactions. The reaction gave the desired product in a fairly low yield (45%). Whilst the yield was slightly low the FT-IR showed all of the characteristic absorptions such as C=O (1697cm-1) and CAr-CAr (1585cm-1). 31P NMR was conducted and showed a sharp singlet at 7.08ppm which indicated full substitution on the phosphazene ring as all of the environments are equivalent. 1H NMR was also conducted and show a singlet at 9.87ppm with an integration of 6H’s which was as expected for the 6 aldehyde protons, two doublets were found in the aromatic region and integrated to 12H’s each which was correct for the number of protons expected. The CHN analysis was almost perfect with less than 0.2% difference from the calculated values, this indicated that the product was of high purity. Mass spectometry also indicated that the desired product had been synthesized as a [M+Na]+ peak was detected at 884.
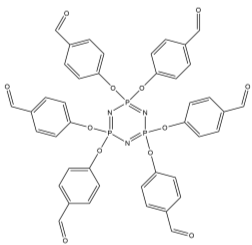
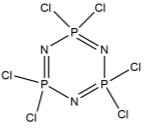

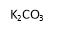
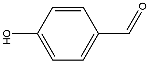
Figure 12 – The reaction scheme for the synthesis of 1.
2.1 Synthesis of Hexalophine and Hexalophine derivatives
2.1.0 Using Diketones
The synthesis of hexalophine and hexalophine derivatives is important due to their porous properties which can be of great importance in fields such as gas storage and catalysis. The method for attaching six lophine units to the ring was a ring condensation of 1 on the six benzaldehyde units attached to the phosphazene ring using a synthesis based upon Radziszewski’s synthesis.1 The product 2 was achieved by reacting 1 along with six equivalents of benzil, ammonium acetate in excess in glacial acetic acid (figure 13). The yield for 2 was quite poor (32%), the FT-IR showed all of the characteristic peaks such as CAr-H (3055cm-1), C=N (1604cm-1) and CAr=CAr (1538cm-1) indicating the desired product had been formed. This was further confirmed by both the 31P and 1H NMR, the 31P NMR showed a singlet at 8.36ppm which indicates full substitution on the phosphazene ring. The 1H NMR showed a singlet at 12.65ppm which had an integration equal to 6H’s which is representative of the 6 imidazole N-H’s, it also showed multiplets in the aromatic region with an integration of 84H’s which is exactly the number of aromatic hydrogens in the desired product. CHN analysis of the product correlated well with the desired structure, especially when three molecules of water were factored into the calculation. It is unsurprising that this compound also contained water due to its porous nature.
 NH4OAc
NH4OAc
Acetic acid
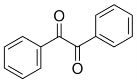
(6eq.)
Figure 13 – The reaction scheme for the synthesis of 2.
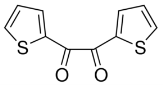
Another reaction designed based upon the ring condensation reaction was the synthesis of 4. This synthesis builds upon the diketone functionality as above, however, in this reaction the R groups of the diketone thenil were thiophene rings as opposed to the phenyl rings of benzil. Thenil like benzil was reacted with 1 and ammonium acetate in glacial acetic acid (figure 14). The yield for this compound, whilst better than 2, was still fairly low (46%). FT-IR showed the characteristic peaks such as CAr-H (3071cm-1), CAr=CAr (1643cm-1) and C=N (1607cm-1) indicating the target molecule had been synthesised. The 31P NMR showed a singlet at 8.26ppm again indicating full substitution. The 1H NMR showed a singlet 12.91ppm with an integration of 6H’s representative of the 6 imidazole N-H hydrogens, it also showed multiplets in the aromatic region with an integration of 60 which was again exactly what was required for the number of aromatic hydrogens, indicating the correct product had been synthesised. The CHN analysis was close to the theorectical values required especially when four water molecules were factored in, again this is unsurprising as this material like 2 is also expected to be porous.
 NH4OAc
NH4OAc
Acetic acid
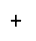
(6eq.)
Figure 14 – The reaction scheme for the synthesis of 4.
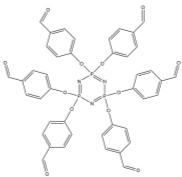
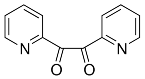
Like thenil and benzil, pyridil is also a diketone where the two R groups are heterocycles, in this case the two heterocycles are pyridine rings. Pyridil was used again with the classical Radziszewski based synthesis to synthesise the compound 5 (Figure 15). The yield for this compound was very low (15%) due to a lot of mass lost upon recrystallization and decomposition upon heating. The FT-IR showed the correct absorptions for the key functional groups such as C=N (1600cm-1) and P=N (1158cm-1) indicating that the target compound had been synthesised. However large OH absorption band (3349cm-1) was also present showing that the product contained a considerable amount of water or ethanol. CHN analysis of the compound showed a vast difference in carbon and nitrogen values when compared to the theoretical values indicating some impurities in the sample. The fact that the product contained considerable amounts solvent would also skew the result of the CHN analysis somewhat.
 NH4OAc
NH4OAc
Acetic acid
(6eq.)
Figure 15 – The reaction scheme for the synthesis of 5.
2.1.1 Using Aldehydes for an alternative preparation
The idea of using aldehydes such as pyridine carboxyaldehyde and benzaldehyde as opposed to the diketones used above was born when questioning on how we could stabilise the pores in the materials we wished to synthesise as larger groups could be added to these aldehyde starting materials. An alternative synthesis of 2 using twelve equivalents of benzaldehyde is illustrated in figure 16.
The reaction was left to reflux for 7 days however the 31P NMR showed a multiplet indicating that full substitution had not occurred or a mix of products was present. Unfortunately, this was a reoccurring theme as the failed synthesis of 3 using twelve equivalents of pyridine carboxyaldehyde (figure 17) also showed a multiplet in the 31P NMR.
 Benzaldehyde (12 eq.)
Benzaldehyde (12 eq.)
Figure 16 – The reaction scheme for and alternative preparation of 2.
 Carboxyaldehyde (12 eq.)
Carboxyaldehyde (12 eq.)
Figure 17 – The reaction scheme for compound 3.
2.2 Oxidation of Hexalophine and its derivatives
The oxidation of hexalophine is of great interest due to the potential properties it may possess, such as it possessing photochromic properties which may lead to light gated pores. For this oxidation an excess of potassium hydroxide was used to create the anion before 50 equivalents of the classical oxidising agent potassium ferrocyanide (III) were used to perform a one electron oxidation to the radical species 6, a yellow to orange colour change is associated with the formation of the radical species. This radical species quickly dimerises and an orange to yellow colour change is associated with this. Upon irradiation with UV light (365nm) the dimer opens to form two radicals, the colour change associated with this was yellow to purple displaying the photochromic behaviour of this compound (figure 18). The radical species is stable for a number of hours in the solid state unless gentle heating is applied which induces dimerization again and the colour change of purple to yellow is associated with this. FT-IR showed characteristic absorbances of CAr-H (2955cm-1), C-N (1301cm-1) and P=N (1198cm-1) which are present in the desired product.
The thiophene hexalophine derivative 4 was also oxidised using the same classical oxidation conditions as above to form its radical species 7. Upon separation the system formed 3 layers, an organic layer, an aqueous layer and a solid substance formed which was soluble in neither. Both the organic layer and solid substance were irradiated with UV light to test for photochromic properties however neither displayed a colour change and thus further testing of this material via UV/Vis spectroscopy is required to determine its nature.

Figure 18 – A RBF containing the hexalophine dimer and radical, the purple is the radical species, the yellow is the dimer species.
3. Conclusions and further work
The aim of this project was to synthesise and characterise functionalised porous organics based upon hexalophine as well as to perform oxidations to the radical species and study their properties such as photochromism. For the most part the aim of the project has been met as two derivatives of hexalophine (4 and 5) and hexalophine (2) itself have been synthesised from simple molecular building blocks and characterised using a range of spectroscopic techniques such as NMR, FT-IR and CHN analysis. The next step in the characterisation of these materials would be to gain crystal structures via x-ray crystallography to show the packing arrangement of these molecules and to prove the existence of pores with in the molecule created from the unique conformation of the phosphazene ring. Additionally, once crystal structures have been gained gas absorption test should be taken to test both the gas uptake properties and selectivity of certain gases. Yields for these experiments were quite low and the aldehyde reactions did not demonstrate full substitution onto the ring, one possible solution would be to try the reaction in a Parr pressure reactor. The oxidation of hexalophine was carried out with great success as the experiment demonstrated the proposed photochromic nature of the material, also the stability of this material with respect to air was very surprising. If possible crystal structures for this material should be obtained in order to investigate further the possibility of light gated pores which when the molecule is in the dimer form are closed but once in the radical form are open.
4. Experimental details
4.0 Materials
All reagents and solvents were used as procured. All NMR data was recorded on a Bruker 400MHz spectrometer and the resulting spectra were processed using ACD Spectrus and Mestrenova. FT-IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR spectrometer. Mass spectrometry was recorded using a Micromass LCT Mass Spectrometer. CHN analysis was carried out by the University of Liverpool analytical services department.
4.1 Synthesis and Charecterisation
C42H30N3O12P3 – 1:
Into a 250ml RBF phosphonitrillic chloride trimer (2.0g, 5.75mmol), 4-hydroxybenzaldehyde (4.21g, 34.5mmol) and THF (100ml) were added along with stirring. To this mixture potassium carbonate (9.5g, 69mmol) was added, the mixture was then heated to reflux for 24 hours. A white precipitate of potassium chloride formed and was filtered off. The filtrate was evaporated to dryness giving a white powder which was the crude product. The crude product was recrystallized in ethyl acetate to give beige needle like crystals of 1 (2.23g, 45%).
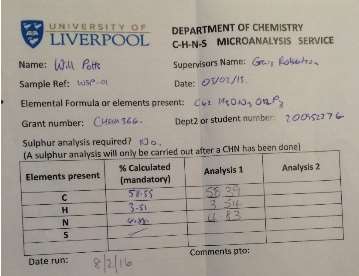
Found C, 58.39; H, 3.54; N, 4.83. C42H30N3O12P3 requires C, 58.55; H, 3.51; N, 4.88. FT-IR vmax/cm-1 1697 (C=O), 1585 (CAr=CAr), 1205 (P=N), 1149 (P=N), 951 (P-O-C). ð›¿H (400MHz; CDCl3; Me4Si) 9.87 (6H, singlet, C(O)-H), 7.68 (12H, doublet, CAr-H), 7.09 (12H, doublet CAr-H); ð›¿P (400MHz; CDCl3;85% H3PO4) 7.08 (3P, singlet, P-O); mp 163°C (from EtOAc); m/z 884 ([M+Na]+), 885.1, 916.1, 917.2.
C126H90N15O6P3 – 2:
Into a 250ml RBF compound 1 (1.01g, 1.17mmol) was added along with glacial acetic acid (50ml), benzil (1.49g, 7.1mmol) before ammonium acetate (3.76g, 48.8mmol) was added along with stirring, the mixture was left to reflux for 3 days. The yellow solution was allowed to cool to room temperature before being neutralised with sodium bicarbonate in ice. The mixture was filtered under vacuum to give the crude product which was recrystallized from ethanol to give 2 (0.75g, 32%).
Found C, 74.47; H, 4.45; N, 10.16. C126H90N15O6P3 requires C, 75.5; H, 4.53; N, 8.96. FT-IR vmax/cm-1 3055 (CAr-H), 1604 (C=N), 1538 (CAr=CAr), 1184 (P=N), 949 (P-O-C). ð›¿H (400MHz; DMSO-d6; Me4Si) 7.19-8.09 (84H, multiplet, CAr-H), 12.65 (6H, singlet, N-H). ð›¿P (400MHz; DMSO-d6;85% H3PO4) 8.36 (3P, singlet, P-O).
C126H90N15O6P3 – 2 Alternative preparation:
Into a 250ml RBF compound 1 (0.5g, 0.58mmol) was added along with benzaldehyde (0.78g, 7.31mmol) and glacial acetic acid (40ml). With stirring ammonium acetate (1.89g, 24.4mmol) was added to the mixture which was left to reflux for seven days. 31P NMR was taken and showed a multiplet indicating the reaction had failed.
C114H78N27O6P3– 3:
Into a 100ml RBF compound 1 (0.5g, 0.58mmol), 4-pyridinecarboxaldehyde (0.75ml, 8mmol) was added along with glacial acetic acid (40ml). With stirring ammonium acetate (1.89g, 24.4mmol) was added to the mixture. The reaction mixture was left to reflux for 5 days. 31P was taken and showed a multiplet indicating that the reaction had failed.
C102H66N15O6P3S12– 4:
Into a 250ml RBF compound 1 (0.5g, 0.58mmol) was added along with glacial acetic acid (35ml), thenil (0.81g, 3.66mmol) before ammonium acetate (1.89g, 24.52mmol) was added along with stirring, the mixture was left to reflux for 4 days. The black reaction mixture was allowed to cool to room temperature before being neutralised with sodium bicarbonate in ice. The mixture was filtered under vacuum to give the crude product which was recrystallized from ethyl acetate and pet ether to give 4 (0.55g, 46%).
Found C, 57.37; H, 3.49; N, 9.41; S, 17.48. C102H66N15O6P3S12requires C, 59.03; H, 3.21; N, 10.12; S, 18.54. FT-IR vmax/cm-1 3071 (CAr-H), 1643 (CAr= CAr), 1607 (C=N), 1159 (P=N), 937 (P-O-C).ð›¿H (400MHz; DMSO-d6; Me4Si) 6.89-8.06 (60H, multiplet, CAr-H), 12.91 (6H, singlet, N-H). ð›¿P (400MHz; DMSO-d6;85% H3PO4) 8.06 (3P, singlet, P-O).
C114H78N27O6P3– 5
Into a 250ml RBF compound 1 (0.55g, 0.62mmol) was added along with 2,2 pyridil (0.71g, 3.88mmol) and glacial acetic acid (40ml). With stirring ammonium acetate (1.99g, 25.8mmol) was added to the mixture which was left to reflux for 24 hours. The green reaction mixture was allowed to cool to room temperature before being neutralised with sodium bicarbonate in ice. The mixture was filtered under vacuum giving the crude product which was recrystallized from ethanol to give 5 (0.19g, 15%).
Found C, 60.88; H, 4.53; N, 12.34 C114H78N27O6P3 requires C, 67.95; H, 3.9; N, 18.77. FT-IR vmax/cm-1 3349 (O-H), 1600 (C=N), 1158 (P=N), 952 (P-O-C).
C126H84N15O6P3 – 6
Into a 250ml RBF compound 2 (0.1g, 0.05mmol) was added along with ethyl acetate (30ml) and water (30ml). With stirring potassium hydroxide (2.0g, 36mmol) was added along with potassium ferrocyanide (0.82g, 2.5mmol). A colour change of yellow to orange and then back to yellow was observed. The biphasic mixture was washed with water (3x50ml) before being evaporated down to dryness and dried under a nitrogen atmosphere. Upon irradiation with UV light of 365nm a colour change of yellow to purple was observed.
FT-IR vmax/cm-1 2955 (CAr-H), 1301 (C-N), 1198 (P=N).
C102H60N15O6P3S12 –7:
Into a 250ml RBF compound 4 (0.1g, 0.05mmol) was added along with ethyl acetate (30ml) and water (30ml). With stirring potassium hydroxide (2.0g, 36mmol) was added along with potassium ferrocyanide (0.82g, 2.5mmol). A colour change of yellow to green and then back to yellow was observed. A 3 phase mixture was observed consisting of an organic layer, an aqueous layer and a solid layer. The solid layer was filtered off and dried. The organic layer was washed with water (3x50ml) before evaporated to dryness. Both materials were treated with UV light of 365nm no colour change occurred.
Acknowledgements:
Dr. C. M. Robertson
Mike Craven
Dr. A. Steiner
References:
1.B. R. Radziszewski, Ber. Dtsch. Chem. Ges., 1877, 10, 70-75.
2.T. Hayashi, K. Maeda and T. Kanaji, Bull. Chem. Soc. Jpn., 1965, 38, 857-+.
3.R. M. Edkins, M. R. Probert, C. M. Robertson, J. A. K. Howard and A. Beeby, RSC Advances, 2014, 4, 5351-5356.
4.Z. L. Zhang, D. D. Yao, T. L. Zhou, H. Y. Zhang and Y. Wang, Chem. Commun. (Cambridge, U. K.), 2011, 47, 7782-7784.
5.T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1965, 38, 685-+.
6.S. Hatano, T. Horino, A. Tokita, T. Oshima and J. Abe, Journal of the American Chemical Society, 2013, 135, 3164-3172.
7.S. Hatano and J. Abe, Phys. Chem. Chem. Phys., 2012, 14, 5855-5860.
8.R. M. Edkins, M. R. Probert, K. Fucke, C. M. Robertson, J. A. K. Howard and A. Beeby, Phys. Chem. Chem. Phys., 2013, 15, 7848-7853.
9.T. Ikezawa, K. Mutoh, Y. Kobayashi and J. Abe, Chem. Commun. (Cambridge, U. K.), 2016, 52, 2465-2468.
10.J. D. Wuest, Chem. Commun. (Cambridge, U. K.), 2005, DOI: 10.1039/b512641j, 5830-5837.
11.J. F. Bickley, R. Bonar-Law, G. T. Lawson, P. I. Richards, F. Rivals, A. Steiner and S. Zacchini, Dalton Trans., 2003, DOI: 10.1039/b212308h, 1235-1244.
12.M. J. Zaworotko, Chem. Commun. (Cambridge, U. K.), 2001, DOI: 10.1039/b007127g, 1-9.
13.J. E. Greedan, J. Mater. Chem., 2001, 11, 37-53.
14.K. Awaga, T. Okuno, A. Yamaguchi, M. Hasegawa, T. Inabe, Y. Maruyama and N. Wada, Physical Review B, 1994, 49, 3975-3981.
15.K. Kanoda and R. Kato, in Annual Review of Condensed Matter Physics, Vol 2, ed. J. S. Langer, Annual Reviews, Palo Alto, 2011, vol. 2, pp. 167-188.
16.T. Isono, H. Kamo, A. Ueda, K. Takahashi, M. Kimata, H. Tajima, S. Tsuchiya, T. Terashima, S. Uji and H. Mori, Phys. Rev. Lett., 2014, 112, 5.
17.P. Sozzani, A. Comotti, R. Simonutti, T. Meersmann, J. W. Logan and A. Pines, Angewandte Chemie-International Edition, 2000, 39, 2695-2698.
18.K. C. Stylianou, J. Rabone, S. Y. Chong, R. Heck, J. Armstrong, P. V. Wiper, K. E. Jelfs, S. Zlatogorsky, J. Bacsa, A. G. McLennan, C. P. Ireland, Y. Z. Khimyak, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Journal of the American Chemical Society, 2012, 134, 20466-20478.
19.P. Brunet, M. Simard and J. D. Wuest, Journal of the American Chemical Society, 1997, 119, 2737-2738.
Appendices:
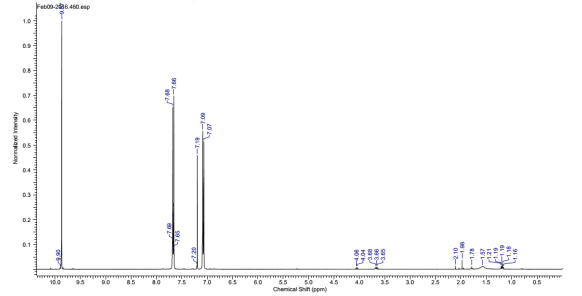 Compound 1:
Compound 1:
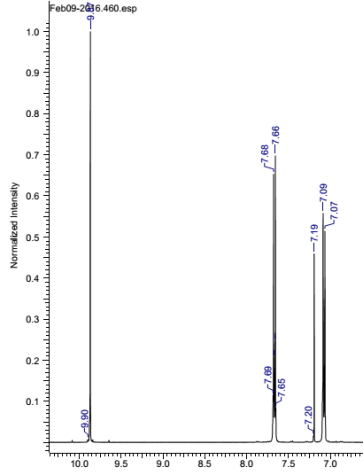
1HNMR
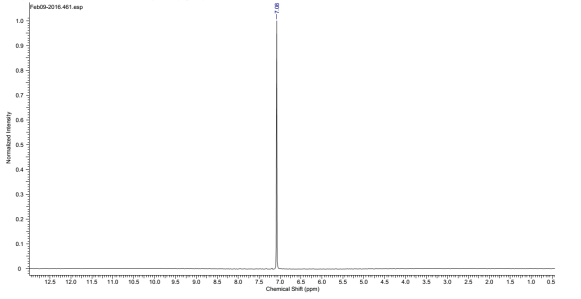
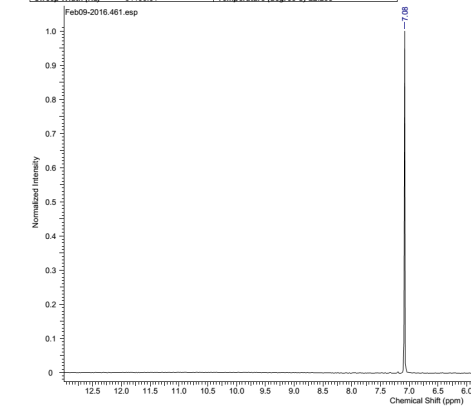
31P NMR
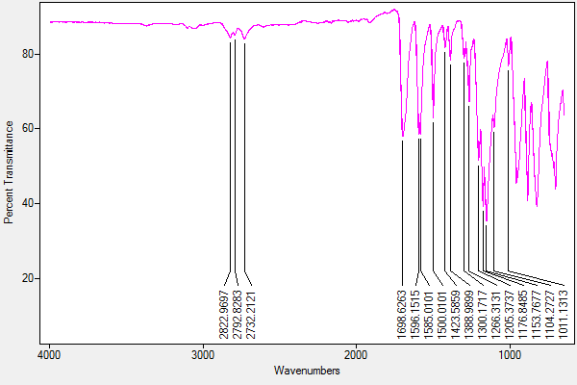
FT-IR Spectrum
CHN analysis
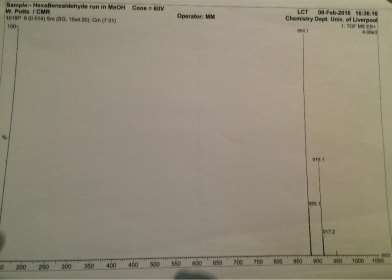
Mass Spec
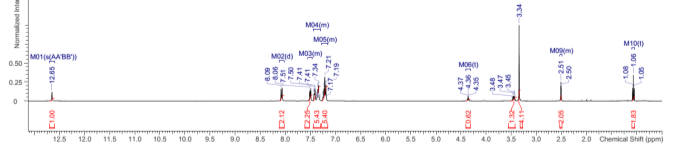 Compound 2:
Compound 2:
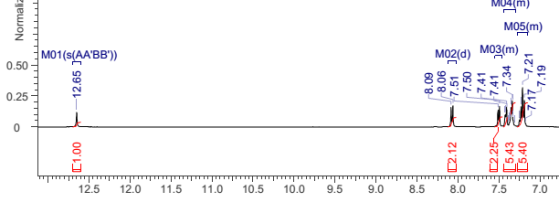 1HNMR
1HNMR
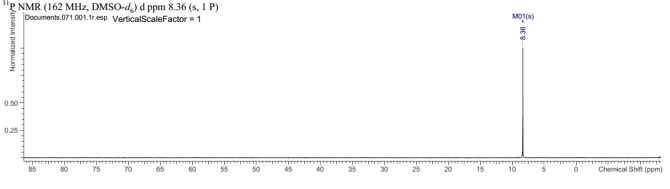
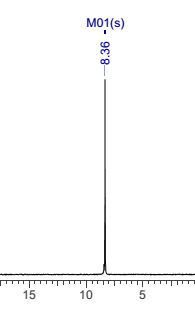
31P NMR
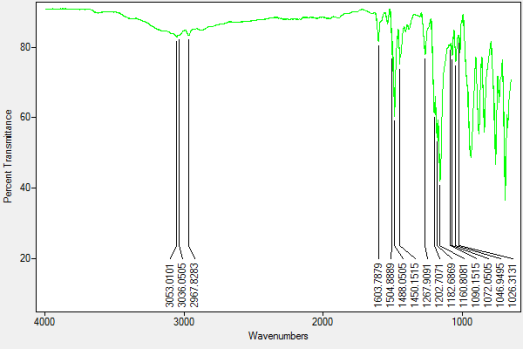
FT-IR
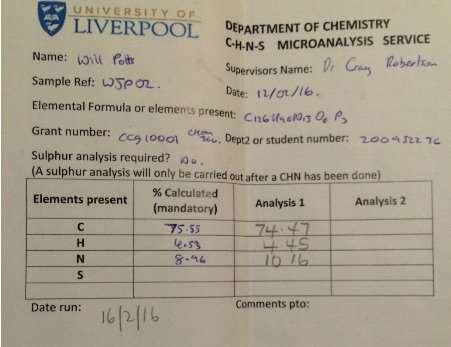
CHN analysis
Compound 4:
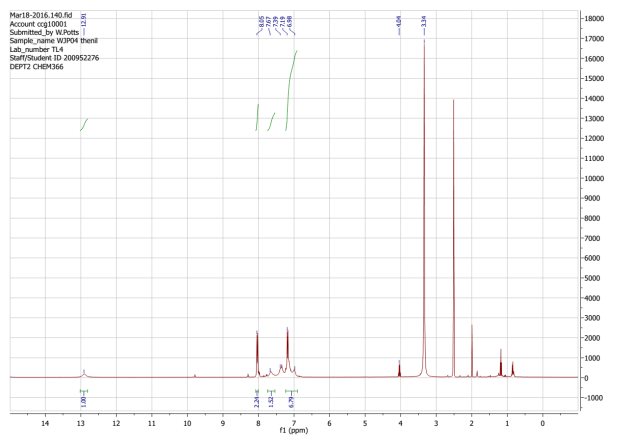
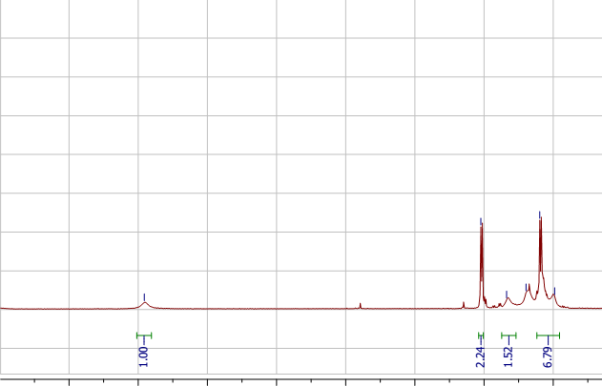
1HNMR
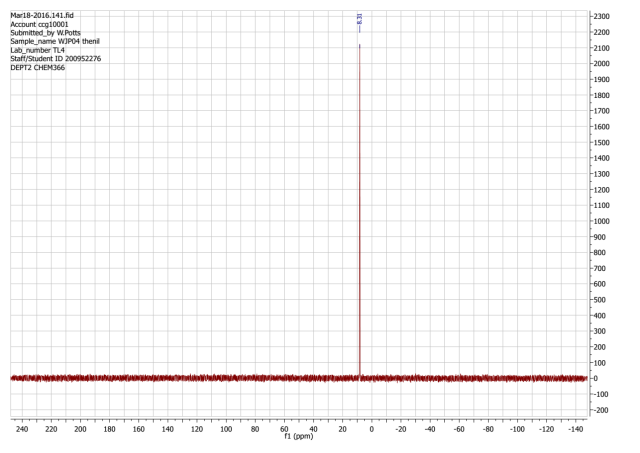
31P NMR

31P NMR
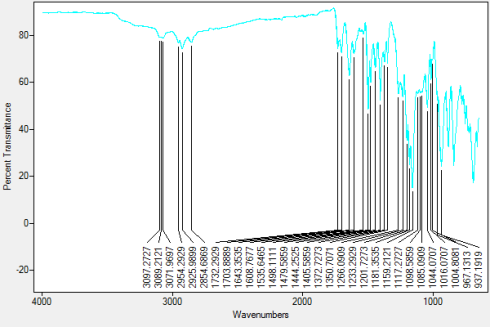
FT-IR
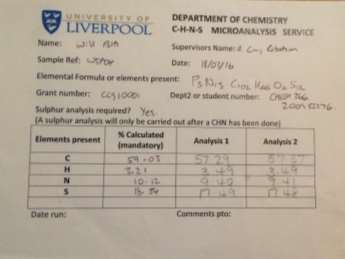
CHN analysis
Compound 5:
FT-IR

CHN analysis
Compound 6:
FT-IR