Nitration of Acetanilide and Methyl Benzoate
Nitration of Acetanilide and Methyl Benzoate by Electrophilic Aromatic Substitution
Abstract
The purpose of this experiment was to synthesize methyl nitro benzoate from methyl benzoate, as well as nitroacetanilide from concentrated nitric acid (HNO3), and concentrated sulfuric acid (H2SO4) by using an electrophilic aromatic substitution reaction. The HNO3 and H2SO4 were combined to form a nitrating solution, which was added to a mixture of methyl benzoate and H2SO4, and the same was done with acetanilide. Following recrystallization, melting point was used to identify and characterize the product of the reaction. The melting point was determined to be 74 ËšC-80 ËšC for methyl nitro benzoate and for nitroacetanilide it was 195 ËšC-200 ËšC, which indicates meta-regiochemistry for methyl benzoate and para-regiochemistry for nitroacetanilide. The percent yield of this reaction for the recrystallized product was 59.3% of methyl nitrobenzoate, while it was 6.75% for nitroacetanilide.
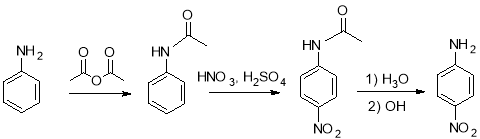
Figure 1: The reaction for the nitration of acetanilide.
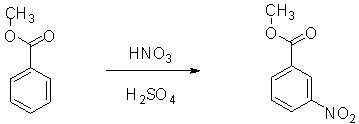
Figure 2: The reaction for the nitration of methyl benzoate.
Experimental
Concentrated sulfuric acid (0.6 mL) and concentrated nitric acid (0.5 mL) were added to a reaction flask and placed in an ice bath. Concentrated sulfuric acid (1 mL) was added to methyl benzoate (0.5 g) in a vial which was then packed in ice, and the same was done with acetanilide. While stirring, the cold H2SO4/HNO3 mixture was added drop-by-drop. After the acid mixture was added, the reaction mixture was removed from the ice to warm to room temperature, with stirring. It was then transferred by Pasteur pipet into a beaker and stirred for five minutes. The methyl benzoate nitration formed white solid, and the acetanilide nitration for a light yellow solid. The crystals were the vacuum filtered with a Buchner funnel. The crude product was recrystallized by adding a distilled water and ethanol slowly while heating the product. While cooling, the solution produced large white crystals for methyl benzoate nitration and light yellow crystals for the acetanilide nitration. The mass, melting point percent yield were obtained.
Results and Discussion
Through the use of electrophilic aromatic substitution, acetanilide is nitrated to nitroacetanilide, while methyl benzoate was nitrated to methyl nitrobezonate. The first step of the reaction involved in the donation of an electron pair, which generates the nitronium ion from nitric acid by protonation and loss of water, using sulphuric acid as the dehydrating agent. The mechanism for methyl benzoate can be seen below.
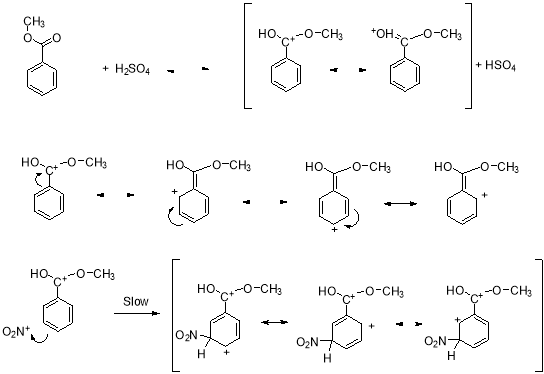
Figure 3: The mechanism of the nitration of methyl benzoate to methyl nitrobenzoate.
To prevent acetanilide from dinitrating, the nitrating solution of HNO3 and H2SO4 were added drop by drop to the acetanilide solution, so that the concentration of the nitrating agent is kept at minimum. The cooler temperatures were used to reduce the reaction rate and help to avoid over nitration.
The electrophilic aromatic substitutions involved the replacement of a proton on an aromatic ring with an electrophile that becomes substituent. The sulfuric acid protonates the methyl benzoate, which creates the resonance stabilized arenium ion intermediate.3 The electron deficient nitronium ion reacts with the protonated intermediate meta position. The ester group is the meta deactivator and the reaction takes place at the meta position because the ortho and para positions are destabilized by adjacent positives charges on the resonance structure.2 The major product of the methyl benzoate nitration is the meta product due to carboxyl and nitro groups both being powerful electron withdrawing groups.
Table 1: The weight, melting point, and percent yield of both Nitroactenilide and Methyl Nitrobenzoate.
|
Product Name |
Crude Weight (g) |
Product Weight (g) |
Percent Yield (%) |
Melting Point (ËšC) |
Literature Melting Point (ËšC) |
|
Nitroactenilide |
0.585 |
0.045 |
6.75% |
195-200 |
214-217 |
|
Methyl Nitrobenzoate |
0.56 |
0.32 |
59.3% |
74-80 |
78-80 |
The actual yield of methyl nitrobenzoate is 0.32 g while the theoretical yield is 0.54 g. The melting point is 74ËšC – 80ËšC, and the value is closed to the literature value which is 78ËšC – 80 ËšC. The percent yield for the methyl benzoate electrophilic aromatic substitution reaction was 59.3% with 0.32g of methyl nitrobenzoate formed. The percent yield for the reaction with acetanilide was 6.75% with 0.045g of nitroacetanilide formed, which can be seen in table 1. The melting point observed was 195-200 ËšC, which can be accounted for impurities in the product, which can be seen below in table 1. Some impurities might be Ortho and Meta directing substances, as well as there could have been some experimental errors that occurred during the experiment such as not overheating solutions during the reactions. These low yields may have resulted from poor recrystallization, product lost during transfer from one apparatus to another, or human error. The missing percent accounts for the impurities removed during recrystallization. However, some product must have been lost in the acetanilide reaction recrystallization because of such a low percent yield.
The melting point of the final product was 74-80 ËšC suggesting that it was formed by meta-substitution. The literature melting point for meta-methyl nitrobenzoate is 78-80 ËšC. Therefore, the melting point is lower than it should be suggesting that an impurity is in the product. This impurity may have occurred due to poor recrystallization or it may have been picked up after recrystallization. The melting point of the nitroacetanilide product was 195-200 ËšC suggesting the para-regiochemistry. The literature melting point for the p-nitroacetanilide is 214-217 ËšC. Therefore, the melting point of the product is a little lower than the literature value, suggesting that an impurity exists in the product from poor recrystallization of contact with an impurity during recrystallization.
The methyl nitrobenzoate product was determined to be meta-substituted based on its melting point range. This can also be proved by evaluating the attack of the benzene ring of methyl benzoate on the electrophililic species and nitric acid.4 The C-OCH3 substituent is a meta-deactivator. Therefore, when the benzene ring attacks the nitronium ion, the NO3+ group will add meta-positon. This creates a resonance stabilized arenium ion without a positive charge on the carbon with the C-OCH3 substituent. Then the proton is removed from the meta position by the weak base, the HSO4-, formed in the creation of the nitronium ion, which reforms the sulfuric acid catalyst.3 Once the proton is removed the substitution product, methyl nitrobenzoate remains.
The nitroacetanilide product was formed by a para-substitution. This can be determined by examining the melting point and comparing it to the literature values for each position.4 However, this can also be determined when examining the electrophilic aromatic substitution reaction of acetanilide on the electrophile, nitric acid. The NHCOCH3 substituent is an ortho-para-activator. Therefore, when the benzene ring of acetanilide attacks the nitronium ion, it can add ortho or para. The para substitution, if more stable than an ortho substitution, will be added because the para position is a further distance from the position of the NHCOCH3 substituent. Therefore, the benzene ring adds at the para position based on the melting point and resonance in the mechanism shown above.
Conclusion
The methyl m-nitrobenzoate and p-nitroacetanilide were prepared. The percentage yield is 6.75% for nitroacetanilide and 59.3% for methyl nitrobenzoate. The melting point of the products are 74ËšC – 80ËšC and 195ËšC – 200ËšC. From the given physical constant, the literature melting point of methyl m-nitrobenzoate is 78 – 80ËšC and 214ËšC – 217ËšC for nitroacetailide, so it can be concluded that the products were methyl m-nitrobenzoate and p-nitroacetanilide.
References
- Wade, Jr., L.G. Organic Chemistry 2003, 722-741.
- Chemistry Laboratory Manual: Susquehanna University 2014, 242-244 & 250
- ChemFinder.Com. Cambridge Soft Corporation. <http://chemfinder.cambridgesoft.com/>.
- Anerjee, Dhruv K. “Ortho and Par % of Key Reaction.” Utkarschemistry. 2013.
Appendix A: Finding the Limiting Reagent
Grams X 1 mol / molecular weight = moles of reactant
Nitric Acid:
0.6 mL X ((1 g/1 mL) X 1 mol) / 98.08 g/mol = 0.0061 mol
Nitric Acid:
0.5 mL X ((1 g/1 mL) X 1 mol) / 63.01 g/mol = 0.0079 mol
Methyl Benzoate:
(0.55 g X 1 mol) / 181.14 g/mol = 0.030 mol
Appendix B: Calculating Theoretical Yield of Methyl Nitrobenzoate
Moles of limiting reagent X molar ratio X molecular weight of product) / 1 mol = theoretical yield
(0.030 X 181.13) / 1 mol = 0.54 g
Appendix C: Calculating Percent Yield
(Actual / theoretical) X 100% = percent yield
(0.32 g/ 0.54 g) X 100% = 59.3%